Extraction Bias in Microbiome Research: Causes, Consequences, and Correction Methods for Accurate Microbial Community Profiling
This article provides a comprehensive analysis of bias introduced during DNA extraction from complex microbial communities, a critical pre-analytical variable that distorts microbiome representation.
Extraction Bias in Microbiome Research: Causes, Consequences, and Correction Methods for Accurate Microbial Community Profiling
Abstract
This article provides a comprehensive analysis of bias introduced during DNA extraction from complex microbial communities, a critical pre-analytical variable that distorts microbiome representation. We explore the foundational causes of bias related to cell lysis efficiency and genomic DNA properties. We detail methodological approaches and kits designed for specific sample types, followed by troubleshooting and optimization strategies to minimize distortion. Finally, we examine validation frameworks and comparative studies benchmarking extraction protocols. This guide equips researchers and drug development professionals with the knowledge to select, optimize, and validate extraction methods, thereby generating more reliable data for translational microbiome science.
Unmasking the Invisible: How DNA Extraction Skews Your View of the Microbial World
Technical Support Center
FAQs & Troubleshooting Guides
Q1: My sequencing data shows very low biomass for Gram-positive bacteria compared to expectations from microscopy. What could be the cause and how can I troubleshoot this?
A: This is a classic symptom of extraction bias favoring Gram-negative cells. Gram-positive bacteria have thicker, more complex peptidoglycan cell walls that are harder to lyse.
- Troubleshooting Steps:
- Verify Lysis Method: Ensure your protocol includes a rigorous mechanical lysis step (e.g., bead beating) in addition to enzymatic (lysozyme) and chemical lysis. Gram-positive cells often require physical disruption.
- Check Bead Beating Parameters: Increase the bead-beating duration or intensity. Use a mix of bead sizes (e.g., 0.1mm and 0.5mm) for more efficient lysis.
- Incorporate Positive Controls: Spike your sample with a known quantity of a Gram-positive control organism (e.g., Bacillus subtilis) and a Gram-negative control (e.g., Escherichia coli) to quantify recovery efficiency.
Q2: I suspect my DNA extraction kit is preferentially recovering DNA from certain taxa, skewing my alpha and beta diversity metrics. How can I test for this?
A: Kit bias is common. To diagnose it, use a mock microbial community with a known, absolute abundance composition.
- Troubleshooting Protocol:
- Acquire a Mock Community: Use a commercially available, sequenced genomic DNA mock community (e.g., from ZymoBIOMICS, ATCC, or BEI Resources).
- Parallel Extraction: Extract DNA from identical aliquots of the mock community using your standard kit and at least two alternative kits/methods known to have different biases.
- Sequencing & Analysis: Sequence all extracts on the same platform and compare the observed proportions to the known "true" proportions. Calculate bias metrics.
Q3: My samples contain both fungi and bacteria, but my ITS sequencing yields are consistently low and variable. How can I improve fungal DNA recovery?
A: Fungal cell walls (chitin) require specialized lysis conditions often omitted from bacterial-focused protocols.
- Troubleshooting Steps:
- Modify Lysis Buffer: Add chitinase or lyticase enzymes to your lysis step to degrade fungal cell walls.
- Extend Lysis Time: Increase incubation time during the enzymatic lysis step (e.g., from 30 min to 1-2 hours at 37°C).
- Optimize Bead Beating: Fungal hyphae and spores are tough. Ensure bead beating includes garnet or silica beads for better physical disruption.
Q4: I am working with soil samples and getting inhibited PCR reactions, leading to failed libraries. How can I address co-extraction of humic substances and PCR inhibitors?
A: Soil is a major source of co-purified inhibitors. Post-extraction purification is critical.
- Troubleshooting Protocol:
- Assess Inhibition: Perform a dilution series PCR (e.g., 1:1, 1:10 template dilution). If amplification improves with dilution, inhibition is confirmed.
- Implement Additional Cleanup: Pass your extracted DNA through a dedicated inhibitor removal column (e.g., Zymo OneStep PCR Inhibitor Removal Kit) or use gel electrophoresis followed by gel excision and purification to separate DNA from small inhibitors.
- Alternative Chemistry: Use a polymerase master mix specifically designed for inhibited samples (e.g., Phusion Blood Direct PCR Master Mix).
Table 1: Comparison of DNA Yield and Community Representation from Three Common Extraction Methods on a ZymoBIOMICS HMRC Mock Community
| Extraction Method | Mean Total DNA Yield (ng) | Gram-Positive:Gram-Negative Ratio (Observed) | Expected G+:G- Ratio | % Recovery of B. subtilis (Gram+) | % Recovery of P. aeruginosa (Gram-) |
|---|---|---|---|---|---|
| Kit A (Enzymatic Lysis Only) | 45.2 ± 5.1 | 0.15 ± 0.03 | 1.0 | 12% ± 3% | 95% ± 8% |
| Kit B (Bead Beating, 2 min) | 68.7 ± 7.3 | 0.82 ± 0.11 | 1.0 | 78% ± 9% | 88% ± 7% |
| Kit C (Bead Beating, 5 min) | 75.1 ± 8.9 | 1.05 ± 0.15 | 1.0 | 102% ± 12% | 81% ± 6% |
Table 2: Impact of Inhibitor Removal Steps on PCR Amplification Success Rate from Complex Fecal Samples
| Purification Step Post-Extraction | PCR Success Rate (16S rRNA Gene) | Mean Cycle Threshold (Ct) Value | A260/A230 Purity Ratio |
|---|---|---|---|
| None (Standard Kit Eluate) | 45% (9/20) | 28.5 ± 3.2 | 1.8 ± 0.4 |
| Silica Column Cleanup | 85% (17/20) | 24.1 ± 2.1 | 2.0 ± 0.2 |
| Size-Selective Gel Purification | 100% (20/20) | 21.8 ± 1.5 | 2.1 ± 0.1 |
Experimental Protocols
Protocol: Benchmarking Extraction Bias Using a Mock Microbial Community
Objective: To quantify the bias introduced by a DNA extraction protocol by comparing sequencing results to a known truth standard.
Materials:
- ZymoBIOMICS HMRC (Log Distribution) Mock Microbial Community.
- Candidate DNA extraction kits (A, B, C).
- Bead beater/homogenizer.
- Qubit Fluorometer and dsDNA HS Assay Kit.
- PCR reagents and 16S/ITS primers.
- NGS library prep kit and sequencer.
Methodology:
- Sample Preparation: Resuspend the mock community standard according to the manufacturer's instructions. Prepare 6 replicate aliquots per extraction method.
- DNA Extraction: Perform extractions on all replicates following each kit's (A, B, C) standard protocol precisely. Include a negative extraction control.
- DNA Quantification: Quantify total DNA yield using fluorometry (Qubit).
- Library Preparation & Sequencing: For each extract, perform targeted amplification of the V4 region of the 16S rRNA gene using dual-indexed primers. Pool libraries in equimolar ratios and sequence on an Illumina MiSeq with 2x250 bp reads.
- Bioinformatic Analysis: Process raw reads through a standardized pipeline (e.g., DADA2 or QIIME 2). Assign taxonomy using a classifier trained on the mock community's reference sequences.
- Bias Calculation: For each taxon in the mock community, calculate: Observed % / Expected %. A value of 1 indicates perfect recovery, <1 indicates under-representation, >1 indicates over-representation.
Protocol: Evaluating Inhibitor Removal Efficiency
Objective: To determine the effectiveness of post-extraction purification steps in removing PCR inhibitors.
Materials:
- Complex sample DNA extracts (e.g., from soil, feces).
- Inhibitor removal columns (e.g., Zymo OneStep PCR Inhibitor Removal Kit).
- Materials for agarose gel electrophoresis and gel extraction.
- PCR master mix, primers, thermocycler.
Methodology:
- Sample Processing: Start with DNA extracted from 10-20 challenging, inhibitor-rich samples.
- Purification: Split each DNA extract into three equal parts:
- Part 1: No further purification.
- Part 2: Purify using an inhibitor removal column. Elute in the same volume as Part 1.
- Part 3: Run on a low-melting point agarose gel. Excise the high-molecular-weight DNA band and purify using a gel extraction kit. Elute in the same volume.
- PCR Amplification: Perform a standardized qPCR assay (e.g., for the 16S rRNA gene) on all purified fractions using identical reaction conditions and template volumes.
- Analysis: Record the PCR success rate (presence of an amplification curve with correct melting temperature) and the Cycle Threshold (Ct) value for each reaction. Compare Ct values and success rates across the three methods.
Visualizations
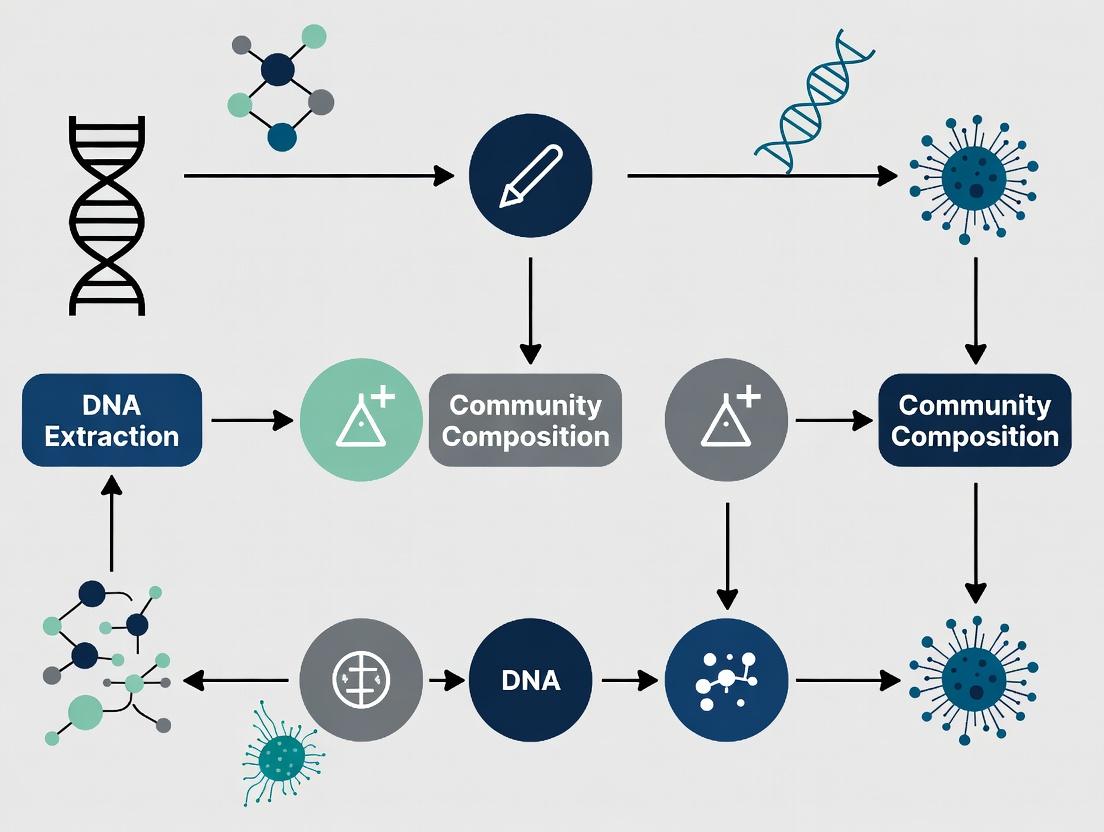
Title: Workflow for Identifying Extraction Bias
Title: Lysis Efficiency by Cell Wall Type
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function & Rationale |
|---|---|
| Mechanical Lysis Beads (0.1mm & 0.5mm mix) | Provides physical shearing force to break robust cell walls (Gram-positive, fungal, spores) that enzymatic lysis alone cannot disrupt. A mix of sizes improves efficiency. |
| Lytic Enzyme Cocktail (Lysozyme, Mutanolysin, Lysostaphin) | Enzymatically degrades specific bonds in bacterial peptidoglycan. A cocktail is crucial for broad-spectrum lysis across diverse Gram-positive taxa. |
| Chitinase or Lyticase | Specific enzymes for digesting chitin in fungal cell walls, essential for improving recovery from eukaryotes in mixed communities. |
| Sequenced Genomic Mock Community (e.g., ZymoBIOMICS HMRC) | A defined mix of microbial genomes with known, absolute abundance. The essential "truth standard" for quantifying extraction and sequencing bias. |
| Inhibitor Removal Columns (e.g., Zymo OneStep) | Removes humic acids, polyphenols, and other co-extracted compounds from complex samples (soil, plants) that inhibit downstream enzymatic steps. |
| PCR Master Mix for Inhibited Samples | Contains polymerases and enhancers resistant to common environmental inhibitors, increasing amplification success rates from difficult samples. |
| Internal DNA Spike-In (e.g., Synthetic dsDNA Oligo) | A known, non-biological DNA sequence added pre-lysis. Allows for absolute quantification and normalization of samples, correcting for variation in extraction efficiency. |
Technical Support Center: Troubleshooting Guide for Lysis Bias in DNA Extraction
This support center is part of a thesis project dedicated to identifying and mitigating bias in microbial community representation introduced during DNA extraction, specifically via differential lysis efficiency.
Frequently Asked Questions (FAQs)
Q1: Why does my extracted DNA underrepresent Firmicutes (Gram-positive) compared to Proteobacteria (Gram-negative) in my mock community sample? A: This is a classic sign of lysis bias. Gram-positive bacteria have a thick, cross-linked peptidoglycan layer that is inherently more resistant to standard lysis protocols (often optimized for Gram-negatives). Without enhanced mechanical or enzymatic disruption, their DNA is not fully released, leading to underrepresentation in downstream sequencing.
Q2: My extracted DNA yields are low. Should I simply increase the bead-beating time? A: Not necessarily. While increasing mechanical disruption can improve Gram-positive lysis, it can simultaneously shear the DNA from already-lysed Gram-negative cells, creating a reverse bias. Excessive bead-beating fragments DNA, making it unsuitable for long-read sequencing. Optimization requires a balanced approach.
Q3: How can I verify if my extraction protocol is introducing lysis bias? A: The gold standard is using a defined mock microbial community with known, even abundances of organisms from different phyla. Extract DNA using your protocol and compare the sequencing results (relative abundances) to the known composition. Significant deviations indicate bias.
Q4: Which is more effective for disrupting tough cells: enzymatic lysis or mechanical lysis? A: They are often used in combination. Mechanical lysis (bead-beating, sonication) is highly effective but shearing. Enzymatic lysis (e.g., lysozyme, mutanolysin for Gram-positives; lysostaphin for Staphylococcus) is gentler and more specific but may be incomplete alone. A sequential enzymatic-mechanical protocol is often optimal.
Quantitative Data on Lysis Efficiency
Table 1: Comparison of DNA Yield from Different Bacterial Phyla Under Standard vs. Enhanced Lysis Protocols Data synthesized from recent studies on mock communities.
| Bacterial Phyla (Example Organism) | Cell Wall Type | Standard Chemical Lysis Yield (ng/µL) | Enhanced Mechanical Lysis Yield (ng/µL) | Yield Increase Factor |
|---|---|---|---|---|
| Escherichia coli (Proteobacteria) | Gram-negative | 45.2 ± 3.1 | 48.5 ± 2.8 | 1.07 |
| Bacillus subtilis (Firmicutes) | Gram-positive | 12.8 ± 2.5 | 38.9 ± 4.2 | 3.04 |
| Micrococcus luteus (Actinobacteria) | Gram-positive (High G+C) | 8.5 ± 1.7 | 35.1 ± 3.7 | 4.13 |
| Bacteroides thetaiotaomicron (Bacteroidetes) | Gram-negative | 40.1 ± 4.0 | 42.3 ± 3.5 | 1.05 |
Table 2: Impact of Bead-Beating Time on DNA Fragment Size and Community Representation
| Bead-Beating Time (min) | Mean DNA Fragment Size (bp) | Observed % Firmicutes | Observed % Proteobacteria |
|---|---|---|---|
| 1.0 | 18,000 | 15% | 78% |
| 3.0 | 8,500 | 32% | 65% |
| 5.0 | 4,200 | 38% | 58% |
| 7.0 | 1,500 | 35% | 45% |
Detailed Experimental Protocols
Protocol 1: Sequential Enzymatic-Mechanical Lysis for Balanced Community Representation This protocol is designed to minimize bias in complex samples.
- Sample Preparation: Resuspend pelleted cells or biomass in 180 µL of enzymatic lysis buffer (20 mM Tris-Cl pH 8.0, 2 mM EDTA, 1.2% Triton X-100).
- Enzymatic Incubation: Add 20 µL of a lysozyme-mutanolysin cocktail (final conc. 20 mg/mL lysozyme, 5 U/µL mutanolysin). Incubate at 37°C for 45 minutes.
- Mechanical Disruption: Transfer the lysate to a tube containing 0.1 mm and 0.5 mm silica/zirconia beads. Process in a high-speed bead beater for 3 cycles of 1 minute each, with 2-minute intervals on ice.
- Inactivation & Purification: Proceed with standard proteinase K/SDS digestion, followed by column-based or magnetic bead purification as per your kit's instructions.
Protocol 2: Mock Community Validation Assay Essential for benchmarking any extraction protocol.
- Acquire Mock Community: Use a commercially available, defined mock community (e.g., ZymoBIOMICS Microbial Community Standard).
- Parallel Extractions: Perform DNA extraction in triplicate using both your standard protocol and a candidate "bias-reduced" protocol (like Protocol 1 above).
- Sequencing & Analysis: Perform 16S rRNA gene amplicon sequencing (V4 region) or shotgun metagenomics on all extracts. Use standard bioinformatics pipelines (QIIME 2, MOTHUR) for taxonomic assignment.
- Bias Calculation: Calculate the relative abundance of each taxon. Compute the fold-deviation from the known, expected abundance. A protocol with lower average fold-deviation across all phyla is less biased.
Visualizations
Title: Workflow of Lysis Bias Impact on Community Profiling
Title: Decision Tree for Selecting a Lysis Protocol
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in Addressing Lysis Bias |
|---|---|
| Lysozyme | Enzyme that hydrolyzes peptidoglycan in Gram-positive cell walls. Essential first step for many tough cells. |
| Mutanolysin | Enzyme that cleaves the glycan strands in peptidoglycan, often used in combination with lysozyme for enhanced activity against Gram-positives. |
| Lysostaphin | Highly specific enzyme that cleaves the pentaglycine bridges in Staphylococcus peptidoglycan. |
| Zirconia/Silica Beads (0.1 mm & 0.5 mm mix) | Small, dense beads for mechanical cell disruption via bead-beating. A mix of sizes improves lysis efficiency across cell types. |
| Defined Mock Microbial Community (e.g., ZymoBIOMICS) | A standardized mixture of known microbial cells used as a positive control to quantitatively measure bias introduced by DNA extraction. |
| Guanidine Thiocyanate Buffer | A potent chemical denaturant that inactivates nucleases and aids in cell lysis, often used in conjunction with mechanical steps. |
| Inhibitor Removal Technology (e.g., PTFE, silica membranes) | Critical for removing humic acids, salts, and other contaminants from complex samples (soil, stool) that can co-purify during harsh lysis. |
Technical Support Center: Troubleshooting Guides & FAQs
Frequently Asked Questions (FAQs)
Q1: Our downstream PCR or sequencing consistently fails after extraction from soil samples. What is the most likely cause and how can we fix it? A1: This is typically caused by co-extraction of humic acids and other phenolic inhibitors. To resolve this:
- Protocol Step: Incorporate a post-lysis purification step using silica-membrane columns specifically designed for inhibitor removal (e.g., with guanidine thiocyanate wash buffers).
- Reagent Solution: Use commercial inhibitor removal kits or add polyvinylpolypyrrolidone (PVPP) to your lysis buffer.
- Validation: Perform a spike-in control with a known quantity of pure DNA from a standard organism to assess inhibition.
Q2: We observe underrepresentation of high-GC content organisms in our metagenomic data. How can we minimize this bias during extraction? A2: GC-bias is often introduced during cell lysis and shearing.
- Protocol Step: Optimize lysis conditions. Use a combination of enzymatic (e.g., lysozyme, mutanolysin) and mild mechanical lysis (e.g., bead-beating for < 2 minutes) instead of harsh chemical or prolonged mechanical methods.
- Protocol Step: For shearing, use adaptive focused acoustics (AFA) or controlled enzymatic fragmentation instead of sonication, which can over-shear AT-rich DNA.
- Validation: Use a mock microbial community with known GC content distribution to benchmark your protocol.
Q3: Our extracted DNA fragments are too short for long-read sequencing platforms. How can we reduce shearing during extraction? A3: Excessive shearing results from aggressive mechanical lysis and improper handling.
- Protocol Step: Reduce bead-beating time and speed. Test vortex adapters vs. dedicated bead beaters.
- Protocol Step: Avoid vigorous pipetting and vortexing of lysates after cells are disrupted.
- Protocol Step: Use agarose plug lysis for particularly shear-sensitive samples.
- Reagent Solution: Add protective agents like EDTA and chromatin-protecting buffers to stabilize DNA post-lysis.
Q4: How do we choose between different DNA extraction kits for a low-biomass, complex sample? A4: Prioritize kits with proven inhibitor removal and bias minimization features. See the comparison table below.
Table 1: Comparison of DNA Extraction Methods on a ZymoBIOMICS Microbial Community Standard
| Extraction Method / Kit | Mean DNA Yield (ng) | Inhibitor Removal Score (1-5) | % Recovery of High-GC Organism* | Post-Extraction Fragment Size (avg. kb) |
|---|---|---|---|---|
| Harsh Bead-Beating (5 min) | 45.2 ± 5.1 | 2 | 15% ± 3% | 4.1 |
| Enzymatic + Mild Beading (1 min) | 38.7 ± 4.3 | 4 | 89% ± 7% | 18.5 |
| Kit A: Soil-Specific | 40.1 ± 3.8 | 5 | 75% ± 5% | 12.3 |
| Kit B: General Stool | 35.5 ± 4.0 | 3 | 65% ± 8% | 8.7 |
| Phenol-Chloroform (Manual) | 50.1 ± 6.5 | 1 | 50% ± 10% | 23.0 |
Based on qPCR and sequencing recovery of *Pseudomonas aeruginosa (67% GC) relative to known input.
Table 2: Impact of Shearing Method on GC-Content Representation
| Shearing Method | Target Size (kb) | % AT-Rich Recovery* | % GC-Rich Recovery* | Size CV (Coefficient of Variation) |
|---|---|---|---|---|
| Probe Sonication | 5 | 100% ± 12% | 62% ± 9% | 35% |
| Bath Sonication | 5 | 110% ± 15% | 58% ± 11% | 45% |
| Adaptive Focused Acoustics (AFA) | 5 | 98% ± 5% | 95% ± 6% | 15% |
| Enzymatic Fragmentation | 5 | 102% ± 4% | 101% ± 5% | 10% |
*Relative recovery compared to unsheared control, measured by qPCR for target genomic regions.
Detailed Experimental Protocols
Protocol 1: Evaluating Co-Extraction Inhibitors
- Extract: Process your sample (e.g., 0.25g soil) using your standard protocol.
- Spike: After extraction, add a known quantity (e.g., 10^6 copies) of a purified, exogenous DNA control (e.g., lambda phage DNA) to an aliquot of the eluted DNA.
- qPCR: Perform qPCR targeting the spike-in DNA in both the spiked sample and a clean buffer control containing the same spike-in amount.
- Calculate: Determine the ΔCq (Cqsample - Cqcontrol). A ΔCq > 2 indicates significant inhibition.
- Troubleshoot: If inhibition is high, repeat extraction with added PVPP (10-20 mg/g sample) or use a kit with enhanced wash steps.
Protocol 2: Benchmarking GC-Bias with a Mock Community
- Standard: Obtain a commercially available mock microbial community with a characterized GC range (e.g., ZymoBIOMICS Microbial Community Standard D6300).
- Extract: Perform DNA extraction using the test and control methods in parallel.
- Quantify: Perform absolute qPCR using strain-specific primers for high-GC and low-GC members.
- Sequence: Perform shallow 16S rRNA gene amplicon or shotgun sequencing.
- Analyze: Calculate the ratio of observed abundance (from qPCR or sequencing reads) to expected abundance for each member. Plot recovery against GC content.
Diagrams
Diagram 1: Sources of Bias in DNA Extraction Workflow
Diagram 2: Strategy for Bias-Minimized Extraction
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in Bias Mitigation |
|---|---|
| Polyvinylpolypyrrolidone (PVPP) | Binds to phenolic compounds and humic acids in lysis buffer, preventing their co-extraction. |
| Guanidine Thiocyanate Buffer | Chaotropic salt used in silica-membrane kits; denatures proteins and helps dissociate inhibitors from DNA. |
| Lysozyme & Mutanolysin | Enzymes for gentle, targeted degradation of bacterial cell walls (Gram+ and Gram-), reducing need for harsh mechanical force. |
| Benchmark Mock Communities | Defined mixes of microbial cells/genomes with known ratios. Essential for quantifying extraction bias (GC, yield, integrity). |
| Adaptive Focused Acoustics (AFA) System | Provides consistent, controllable mechanical energy for cell lysis and DNA shearing, minimizing fragment size variability and bias. |
| Size-Selective Magnetic Beads | Allow for selection of optimal fragment sizes post-extraction, removing overly sheared DNA that may skew composition. |
| PCR Inhibitor Removal Beads | Specific magnetic beads (e.g., Sera-Mag) coated with polymers that bind inhibitors, used as a post-extraction clean-up. |
FAQs & Troubleshooting Guides
Q1: My alpha diversity metrics (e.g., Shannon, Chao1) show significant differences between sample groups, but I suspect extraction bias. How can I troubleshoot this? A: Spurious alpha diversity differences are a primary symptom of extraction bias. Follow this protocol:
- Internal Standard Spike-In: Prior to extraction, add a known quantity of synthetic cells (e.g., Pseudomonas fluorescens strain not found in your samples) or DNA beads (e.g., from ZymoBIOMICS Spiked-in Standards) to each sample.
- Quantitative PCR (qPCR): Post-sequencing, quantify the recovered reads from your spike-in across all samples using a specific qPCR assay.
- Data Normalization: Calculate the recovery efficiency. Use this to normalize your community sequencing data (e.g., by scaling counts proportional to spike-in recovery) before re-calculating diversity metrics.
Q2: My PERMANOVA results for beta diversity are significant, but my positive controls (identical mock communities) cluster separately. What's wrong? A: This indicates your extraction protocol differentially lyses community members, distorting true biological distances.
- Troubleshooting Step: Implement a standardized Mock Community Challenge.
- Obtain commercially defined mock communities (e.g., ZymoBIOMICS Microbial Community Standard, ATCC Mock Microbial Communities).
- Subject them to your DNA extraction protocol alongside your samples.
- Analyze the resulting composition via 16S rRNA gene or shotgun sequencing.
- Compare the observed vs. expected abundance. Significant deviations confirm protocol-induced bias.
Q3: My differential abundance analysis (e.g., DESeq2, LEfSe) flags many taxa, but I am concerned about false positives from extraction inefficiency. How do I verify? A: Extraction bias can create false differential abundance. Verification requires a multi-protocol approach.
- Protocol: Cross-Validation with Complementary Lysis Methods
- Split each sample into three aliquots.
- Apply three different lysis protocols: a) Mechanical Bead Beating (gold standard for tough cells), b) Enzymatic Lysis (gentle, Gram-negative biased), c) A commercial kit known for your sample type.
- Perform library preparation and sequencing under identical conditions.
- Perform differential abundance analysis on each dataset separately. Taxa consistently identified as significant across all protocols are more likely to be true biological signals, not extraction artifacts.
Q4: My candidate biomarker (a specific bacterial genus) for a disease state is strongly significant but has known tough cell walls. Could it be an artifact? A: Yes. Hard-to-lyse organisms (e.g., Mycobacteria, Gram-positive bacilli) can be systematically under-represented in control groups if cases are processed differently, creating a false biomarker.
- Mitigation Protocol:
- Microscopy Correlation: Perform fluorescence in situ hybridization (FISH) or calcofluor white staining on sample aliquots to visually confirm the physical presence/absence of the candidate biomarker.
- Spike-In Recovery Test: As in Q1, but use a hard-to-lyse surrogate cell spike-in relevant to your candidate (e.g., Bacillus subtilis spores for a Gram-positive biomarker). Low and variable recovery confirms a bias problem.
- Protocol Optimization: Re-extract samples using a protocol with enhanced mechanical lysis (e.g., extended bead-beating time, use of smaller beads) and re-run the biomarker discovery pipeline.
Research Reagent Solutions Toolkit
| Item | Function & Rationale |
|---|---|
| Defined Mock Communities (e.g., ZymoBIOMICS, ATCC MSA-1003) | Provides a "truth set" of known composition to benchmark extraction bias, accuracy, and reproducibility. |
| Exogenous Spike-Ins (e.g., SynDNA, cultured exotic cells, SIRVs for metatranscriptomics) | Controls for variability in lysis efficiency, DNA recovery, PCR inhibition, and normalization between samples. |
| Inhibitor Removal Beads/Columns (e.g., Zymo OneStep PCR Inhibitor Removal, Qiagen PowerClean) | Removes humic acids, phenols, and other co-extracted compounds that inhibit downstream enzymatic steps, reducing bias. |
| Standardized Bead Beating Tubes (e.g., Lysing Matrix E, garnet/silica bead mixes) | Ensures consistent mechanical shearing force across samples to improve lysis uniformity of diverse cell types. |
| DNA/RNA Shield or similar preservation buffer | Immediately inactivates nucleases and stabilizes community composition at collection, preventing shifts prior to extraction. |
Data Summary: Impact of Lysis Method on Observed Composition
Table 1: Recovery of Taxa from a Defined Mock Community Using Different Lysis Protocols.
| Taxon (Cell Wall Type) | Expected % Abundance | Observed %: Bead Beating | Observed %: Enzymatic Lysis Only | Observed %: Thermal Lysis |
|---|---|---|---|---|
| Pseudomonas aeruginosa (Gram-negative) | 25% | 26% (±2%) | 38% (±5%) | 22% (±8%) |
| Escherichia coli (Gram-negative) | 25% | 24% (±1%) | 35% (±4%) | 20% (±7%) |
| Staphylococcus aureus (Gram-positive) | 25% | 25% (±3%) | 12% (±6%) | 8% (±4%) |
| Enterococcus faecalis (Gram-positive) | 25% | 25% (±2%) | 15% (±5%) | 10% (±3%) |
Data is illustrative. Bias magnitude depends on specific protocols. Bead beating shows highest fidelity.
Experimental Protocol: Standardized Extraction Bias Audit
Title: Protocol for Auditing DNA Extraction Bias in Microbial Community Studies. Objective: To quantify protocol-induced bias in microbial community representation. Steps:
- Sample Preparation: Aliquot identical volumes of your typical homogenized sample (e.g., stool, soil slurry). Include replicates.
- Spike-In Addition: To each aliquot, add a consistent volume of a combined spike-in solution containing:
- Soft-cell spike: Known count of Salmonella enterica cells.
- Hard-cell spike: Known count of Bacillus subtilis spores.
- SynDNA spike: Known quantity of synthetic DNA oligonucleotides (non-competitive).
- Parallel Extraction: Extract DNA from all aliquots using: a) Your standard protocol, b) A benchmark harsh mechanical protocol (e.g., prolonged bead beating), c) A commercial kit optimized for your matrix.
- Quantitative Analysis:
- Perform absolute qPCR for each spike-in target to calculate recovery efficiency.
- Perform high-throughput sequencing (16S/ITS/shotgun) on all extracts.
- Bias Calculation:
- For sequencing data: Compare the relative abundance profiles of the endogenous taxa across protocols using PERMANOVA.
- For spike-ins: Calculate % recovery for each. High variance or low recovery of hard-cell spikes indicates significant bias.
Diagram Title: How Extraction Bias Distorts Downstream Results
Diagram Title: DNA Extraction Bias Audit Protocol Workflow
Troubleshooting & FAQ Center for Microbial DNA Extraction & Bias
This support center addresses common experimental challenges in microbiome studies, framed within the thesis of addressing bias in microbial community representation during DNA extraction research.
FAQ: Addressing Bias and Contamination
Q1: My 16S rRNA sequencing results from gut samples consistently show high levels of Lactobacillus, but qPCR validation suggests they are a minor component. What could cause this discrepancy? A: This is a classic sign of lysis bias during DNA extraction. Gram-positive bacteria like Lactobacillus have thick peptidoglycan layers and are more resistant to mechanical lysis compared to Gram-negative bacteria. If your protocol is not optimized for robust mechanical disruption (e.g., bead beating), you will under-extract Gram-positives, making the relative abundance of well-lysed Gram-negatives appear artificially high in downstream sequencing. Lactobacillus may lyse efficiently in your protocol, making its abundance seem inflated relative to other, unlysed Gram-positives. Implement a standardized bead-beating step and validate with a mock community containing known ratios of Gram-positive and Gram-negative cells.
Q2: In skin swab studies, we detect high amounts of soil-associated Actinobacteria in negative controls. What is the source and how can we mitigate it? A: This is likely reagent contamination. Many DNA extraction kits and PCR reagents contain trace environmental bacterial DNA, often from Actinobacteria like Propionibacterium (now Cutibacterium), which are common in fermentation processes used to manufacture enzymes. This bias severely impacts low-biomass samples like skin swabs.
- Solution: Always process extraction and PCR negative controls in parallel. Use dedicated, UV-irradiated laminar flow hoods for low-biomass work. Consider using ultrapure, microbiome-certified reagents. Employ bioinformatic tools like
decontam(R package) to identify and remove contaminant sequences based on their prevalence in negative controls.
Q3: When comparing microbial diversity in soil samples, how do we account for bias introduced by co-extracted humic acids and other inhibitors? A: Co-extraction of inhibitors is a major bias leading to false negatives in PCR amplification. Humic acids inhibit polymerase activity, but not uniformly, creating a bias in which microbial taxa are amplified and sequenced.
- Solution: Include a post-extraction purification step using silica columns or chemical flocculation specifically designed for humic acid removal (e.g., with polyvinylpolypyrrolidone (PVPP)). Quantify inhibition using a spike-in control (e.g., known quantity of a foreign DNA sequence) added to the PCR reaction and measure its amplification efficiency compared to a clean control.
Q4: Our meta-analysis of publicly available gut microbiome datasets shows high variability in the reported abundance of Methanobrevibacter. Could extraction bias be a factor? A: Absolutely. Archaea like Methanobrevibacter have distinct cell wall compositions (pseudopeptidoglycan) that make them susceptible to lysis bias. Studies using harsh mechanical lysis will report higher archaeal abundance than those using only enzymatic lysis. This methodological bias makes cross-study comparisons invalid without standardization.
- Solution: When designing new studies or re-analyzing data, always note the DNA extraction protocol used. For future experiments, use a protocol validated on mock communities containing archaea. For meta-analyses, only compare studies using highly similar extraction methods or use batch-correction statistical tools.
Table 1: Comparative Lysis Efficiency and Bias Across Cell Types
| Cell Type / Structure | Example Taxa | Susceptibility to Enzymatic Lysis | Susceptibility to Mechanical Bead Beating | Risk of Bias Without Bead Beating |
|---|---|---|---|---|
| Gram-negative bacteria | Escherichia, Bacteroides | High | High (Potential over-lysis) | Under-representation of robust cells |
| Gram-positive bacteria | Firmicutes, Lactobacillus | Low | High | Severe Under-representation |
| Mycobacteria (waxy cell walls) | Mycobacterium | Very Low | Very High Required | Near-total absence |
| Archaea | Methanobrevibacter | Low | Medium-High | Under-representation |
| Yeasts/Fungi | Candida, Saccharomyces | Low | High | Severe Under-representation |
| Spores | Clostridium spores | Very Low | Very High Required | Near-total absence |
Table 2: Common Sources of Contaminating DNA in Low-Biomass Studies
| Source | Common Contaminant Taxa Identified | Primary Impacted Sample Type | Mitigation Strategy |
|---|---|---|---|
| DNA Extraction Kits | Pseudomonas, Delftia, Sphingomonas, Burkholderia | Skin, placenta, lung, tissue | Use kit-negative controls; choose low-biomass validated kits |
| PCR Reagents (Polymerase) | Propionibacterium (Cutibacterium), Bradyrhizobium | All low-biomass samples | Use ultrapure, amplified DNA-free reagents |
| Laboratory Water | Varied environmental bacteria | All samples | Use certified DNA-free, UV-treated water |
| Laboratory Surfaces & Air | Human skin flora (Staphylococcus, Corynebacterium), Environ. | Low-biomass environmental swabs | UV irradiation, dedicated clean rooms, filter tips |
Detailed Experimental Protocol: Standardized Bead-Beating for Comprehensive Lysis
Objective: To minimize lysis bias and achieve representative DNA extraction from diverse microbial cells in complex communities (e.g., stool, soil).
Materials:
- Sample (e.g., 180-220 mg stool, 250 mg soil)
- Lysis buffer (e.g., containing guanidine thiocyanate, Tris, EDTA)
- Proteinase K
- Acid-washed, sterile zirconia/silica beads (mix of 0.1mm and 0.5mm)
- Bead-beating homogenizer (e.g., FastPrep-24, TissueLyser II)
- Phenol:Chloroform:Isoamyl Alcohol (25:24:1)
- Isopropanol and Ethanol (70%)
- Silica membrane spin columns
- Elution buffer (10 mM Tris-HCl, pH 8.5)
Method:
- Homogenization: Transfer sample to a 2ml bead-beating tube containing ~500μl of lysis buffer and 100-500μl of bead mix.
- Mechanical Disruption: Secure tubes in a bead-beater. Process at 6.5 m/s for 60 seconds. For extremely tough spores or fungi, a second cycle may be necessary. Cool samples on ice for 2 mins between cycles to prevent DNA shearing.
- Enzymatic Lysis: Add Proteinase K (final conc. ~2 mg/ml). Incubate at 56°C for 30-60 minutes with agitation.
- Inhibitor Removal: For environmental samples, add PVPP (5% w/v) to the lysate, vortex, and incubate on ice for 10 minutes before centrifugation.
- Nucleic Acid Separation: Centrifuge at 12,000 x g for 5 min. Transfer supernatant to a new tube. Add an equal volume of Phenol:Chloroform:Isoamyl Alcohol, vortex, centrifuge. Transfer the upper aqueous phase.
- DNA Binding & Washing: Add 0.7 volumes of isopropanol, mix, and load onto a silica column. Centrifuge, wash with 70% ethanol, and dry the membrane.
- Elution: Elute DNA with 50-100μl of pre-warmed elution buffer.
Validation: Spike the sample with a known quantity of an exogenous control (e.g., cells from Pseudomonas syringae, not typically found in gut/soil) before lysis. Use qPCR specific to this control to calculate absolute recovery efficiency.
Visualizations
Title: Workflow for Bias-Aware DNA Extraction
Title: Contamination Pathways in Low-Biomass Studies
The Scientist's Toolkit: Essential Research Reagents & Materials
| Item | Function & Role in Reducing Bias |
|---|---|
| Zirconia/Silica Beads (0.1mm & 0.5mm mix) | Maximizes mechanical shearing force for robust cell wall disruption of Gram-positives, spores, fungi. |
| Mock Microbial Communities (e.g., ZymoBIOMICS) | Contains defined ratios of Gram-positive, Gram-negative, and fungal cells. Essential for validating extraction bias and sequencing pipeline accuracy. |
| Polyvinylpolypyrrolidone (PVPP) | Binds to and precipitates polyphenolic inhibitors (e.g., humic acids) from environmental samples, reducing PCR inhibition bias. |
| UltraPure DNase/RNase-Free Water | Minimizes introduction of contaminating environmental DNA, critical for low-biomass and negative control preparations. |
| Microbiome Certified DNA Extraction Kits | Kits specifically tested for low contaminant DNA background and validated on diverse cell types. |
| Exogenous Spike-in Control DNA (e.g., pBR322) | Added pre-extraction to quantify absolute DNA recovery efficiency; added pre-PCR to quantify inhibition. |
| DNA LoBind Tubes | Reduces adsorption of low-concentration DNA to tube walls, preventing loss bias. |
Choosing Your Weapon: A Guide to Bias-Aware DNA Extraction Protocols and Kits
Technical Support Center
Troubleshooting Guides & FAQs
Q1: My downstream 16S rRNA gene sequencing shows strong bias towards Gram-negative bacteria, even when I know Gram-positives are present. Which lysis step is most likely the issue? A: This is a classic sign of incomplete lysis of Gram-positive bacteria, which have thicker, more complex peptidoglycan cell walls. Mechanical lysis (e.g., bead beating) is often required. If you used only enzymatic (e.g., lysozyme) or chemical (e.g., SDS) methods, they may be insufficient. Solution: Incorporate a mechanical disruption step, either alone or in a hybrid protocol. For sensitive communities, use short, controlled bead-beating cycles (e.g., 2x 45 seconds with cooling) to prevent DNA shearing while improving Gram-positive recovery.
Q2: I am getting very low DNA yield after using a harsh bead-beating protocol. What went wrong? A: Excessive mechanical force can shear genomic DNA into fragments too small for efficient precipitation or binding to spin columns, leading to low yields and loss of high-molecular-weight DNA. Solution: Optimize bead-beating parameters. Use smaller beads (0.1mm) for bacteria, larger for fungi. Reduce beating time or speed. Implement a cooling step (ice bath between cycles). Consider a follow-up gentle enzymatic step (e.g., proteinase K) to release sheared DNA from cellular debris.
Q3: My extracted DNA appears to have inhibitors for PCR, even after cleanup. Could the lysis reagents be the cause? A: Yes. Chemical lysis agents like CTAB or SDS, if not adequately removed during subsequent purification, can inhibit enzymatic reactions. Solution: Ensure proper precipitation and washing steps. For CTAB protocols, use multiple chloroform:isoamyl alcohol washes. Consider switching to inhibitor-removal specific spin columns. Alternatively, dilute the DNA template in the PCR reaction.
Q4: When I combine lysozyme incubation with bead beating, my community profile shifts. How do I balance these steps? A: Order and duration matter. Prolonged enzymatic pre-treatment can lead to autolysis of some species and alter the "snapshot" of the community. Solution: Standardize incubation times precisely. Consider a shorter, milder enzymatic pre-treatment (e.g., 37°C for 15-30 min) to weaken cell walls, followed by a shorter, gentler bead-beating step. This combination can improve lysis efficiency while minimizing bias from differential autolysis.
Q5: For soil samples, I get inconsistent results between replicates. How can I improve lysis homogeneity? A: Soil particles can protect cells and cause uneven lysis. Solution: Ensure thorough sample homogenization before lysis. Increase the sample aliquot size to improve representativeness. Use a consistent and vigorous mechanical lysis method (e.g., bead beating) for all replicates. A combination approach is often best: chemical lysis (e.g., hot SDS-based buffer) to dissociate cells from particles, followed by mechanical disruption.
Comparative Data Table: Lysis Method Characteristics
| Lysis Method | Typical Agents/Devices | Optimal For | Risk of Bias | DNA Fragment Size | Inhibitor Co-extraction Risk |
|---|---|---|---|---|---|
| Mechanical | Bead mill, French press, Sonication | Gram-positives, Spores, Environmental samples (soil) | Low (if optimized) - provides broad lysis | Can be short (<10 kb) if harsh | Moderate (from disrupted particles) |
| Enzymatic | Lysozyme, Proteinase K, Mutanolysin | Gram-negatives, Cultured cells, Sensitive applications | High (species-specific enzyme efficacy) | Large (>50 kb) | Low |
| Chemical | SDS, CTAB, Alkaline, Detergents | Gram-negatives, Simple protocols, High-throughput | High (differential wall susceptibility) | Large (>20 kb) | High (requires clean-up) |
| Combined | e.g., Lysozyme + Beads + SDS | Complex communities, Robust representation | Lowest when strategically sequenced | Variable (can be optimized) | Moderate-High |
Experimental Protocol: Hybrid Lysis for Minimal Community Bias
Objective: To extract microbial community DNA from stool/soil with reduced compositional bias. Reagent Solutions:
- Lysis Buffer: 100 mM Tris-HCl (pH 8.0), 100 mM EDTA (pH 8.0), 1.5 M NaCl, 2% (w/v) CTAB, 2% (w/v) SDS.
- Enzymes: Lysozyme (50 mg/mL stock), Proteinase K (20 mg/mL stock).
- Mechanical: 0.1 mm and 0.5 mm zirconia/silica beads.
- Inhibitor Removal: Polyvinylpolypyrrolidone (PVPP).
Procedure:
- Weigh 0.25 g of sample into a sterile, bead-beating tube.
- Add 100 µL of lysozyme solution (final conc. 5 mg/mL). Vortex. Incubate at 37°C for 15 minutes.
- Add 750 µL of pre-warmed (60°C) Lysis Buffer and 50 mg of a 1:1 mix of 0.1mm and 0.5mm beads. Add ~10 mg PVPP.
- Mechanical Lysis: Secure tubes on a bead beater. Process at 5.5 m/s for 2 cycles of 45 seconds each, with samples placed on ice for 2 minutes between cycles.
- Add 20 µL of Proteinase K (final conc. 0.4 mg/mL). Mix by inversion.
- Incubate at 55°C for 30 minutes with gentle agitation.
- Proceed with standard phenol-chloroform extraction or column-based purification.
Strategic Lysis Decision Workflow
Research Reagent Solutions Toolkit
| Reagent / Material | Primary Function | Consideration for Bias Mitigation |
|---|---|---|
| Zirconia/Silica Beads (0.1 mm) | Mechanical shearing of tough cell walls (Gram-positives). | Essential for lysis of hardy cells; small size increases shear force. |
| CTAB (Cetyltrimethylammonium bromide) | Chemical lysis & polysaccharide removal. | Critical for soil/plant samples to remove humic acids (PCR inhibitors). |
| Lysozyme | Enzymatic hydrolysis of peptidoglycan in bacterial cell walls. | More effective on Gram-positives; pre-treatment can reduce harsh beating needed. |
| Proteinase K | Enzymatic digestion of proteins & inactivation of nucleases. | Used post-mechanical lysis to liberate DNA from debris and protect it. |
| PVPP | Binds and removes phenolic compounds. | Reduces co-extraction of inhibitors from environmental samples. |
| Inhibitor Removal Microcolumns | Silica-membrane based purification of DNA. | Critical step after chemical lysis to remove residual SDS/CTAB that inhibit PCR. |
Troubleshooting Guides & FAQs
Q1: My stool DNA extraction yields consistently low DNA concentration. What are the primary causes and solutions? A: This is often due to incomplete cell lysis of robust Gram-positive bacteria or inhibitor carryover. First, increase the bead-beating step duration to 4-5 minutes and use a smaller (e.g., 0.1mm) bead size for more effective mechanical disruption. Second, incorporate a specialized inhibitor removal step, such as using polyvinylpolypyrrolidone (PVPP) columns or adding bovine serum albumin (BSA) to downstream PCR reactions to neutralize phenolic compounds.
Q2: How do I mitigate the issue of "humic acid contamination" in soil DNA that inhibits downstream PCR? A: Humic acids co-precipitate with DNA. Use a post-lysis purification protocol. After initial lysis, add 120 µl of 5M potassium acetate (pH 4.8), incubate on ice for 15 minutes, then centrifuge. This selectively precipitates humics. Follow with a silica-column or CTAB-based purification. For severe contamination, commercial "Humic Acid Removal" spin columns are effective.
Q3: When extracting from biofilms, my protocol seems to bias against extracellular polymeric substance (EPS)-embedded cells. How can I improve representation? A: The EPS matrix physically shields cells. Implement a pre-treatment step: gently resuspend the biofilm sample in 1X phosphate-buffered saline (PBS) with 1 mM dithiothreitol (DTT) or 10 mM sodium metaperiodate. Incubate at room temperature for 30 minutes with mild agitation. This reduces disulfide bonds and breaks polysaccharide cross-links without lysing cells, allowing more uniform access to lysis reagents.
Q4: For tissue samples, host DNA vastly overshadows microbial DNA. What are the best practices to enrich for microbial biomass? A: Perform differential lysis and centrifugation. First, homogenize tissue in a gentle, non-ionic detergent lysis buffer (e.g., with 1% Triton X-100). Centrifuge at low speed (500 x g) to pellet host nuclei and debris. Transfer the supernatant, which is enriched for microbial cells, to a new tube. Pellet these cells at high speed (10,000 x g) and proceed with a standard microbial DNA extraction protocol on this pellet.
Q5: My extraction replicates from the same sample type show high variability in community alpha diversity. Is this technical noise, and how can I reduce it? A: This is often technical bias from inconsistent lysis. Standardize the homogenization step rigorously. For stool, soil, and biofilm, use a standardized bead-beating time, bead type/size, and power setting. For tissues, ensure consistent homogenization time and speed. Always process samples in the same bead-beating tube type, as tube geometry affects lysis efficiency. Include a homogenized "mock community" control in each extraction batch to quantify this technical variability.
Key Experimental Protocols
Protocol 1: Modified MOBIO PowerSoil Pro Kit Protocol for Inhibitor-Rich Soils.
- Weigh 0.25 g of soil into a PowerBead Pro tube.
- Add 60 µl of solution C1 (modified to contain 2% w/v PVPP).
- Heat at 65°C for 10 minutes.
- Secure tubes on a vortex adapter and vortex horizontally at maximum speed for 15 minutes.
- Centrifuge at 10,000 x g for 1 minute.
- Transfer supernatant to a clean tube. Add 250 µl of solution C2 and 200 µl of 5M potassium acetate (pH 4.8). Vortex, incubate on ice for 15 min, centrifuge at 15,000 x g for 10 min.
- Transfer 600 µl of supernatant to a new tube, add 200 µl of solution C3, vortex, and incubate on ice for 5 min.
- Centrifuge at 15,000 x g for 10 min. Load all supernatant onto an MB Spin Column.
- Complete the protocol as per manufacturer's wash and elution steps.
Protocol 2: Enzymatic + Mechanical Lysis for Robust Biofilms.
- Scrape and suspend biofilm in 1 ml TE buffer.
- Pre-treatment: Add DTT to 1 mM final concentration. Incubate 30 min at 37°C with gentle rotation.
- Enzymatic Lysis: Add Lysozyme (final 10 mg/ml) and Mutanolysin (final 100 U/ml). Incubate 1 hr at 37°C.
- Chemical Lysis: Add SDS to 1% final and Proteinase K to 0.5 mg/ml. Incubate 1 hr at 56°C.
- Mechanical Lysis: Transfer lysate to a tube with 0.1mm zirconia beads. Bead-beat for 3 x 1 min pulses, cooling on ice between pulses.
- Proceed with phenol-chloroform extraction or column purification.
Data Presentation: Protocol Selection Matrix
Table 1: Recommended Primary Lysis Method by Sample Type
| Sample Type | Dominant Challenge | Recommended Primary Lysis | Bead-Beating Intensity | Key Inhibitor Removal Step | Typical Yield Range (ng DNA/g) |
|---|---|---|---|---|---|
| Stool | Variable composition, inhibitors (bile salts) | Bead-beating + Chemical | High (3-5 min) | PVPP column or BSA in PCR | 1,000 - 10,000 |
| Soil | Humic acids, diverse cell walls | Bead-beating + Chemical | Very High (5-10 min) | Potassium acetate/CTAB precipitation | 100 - 5,000 |
| Biofilm | EPS matrix, aggregated cells | Enzymatic pre-treatment + Bead-beating | Medium-High (2-4 min) | Calcium chloride precipitation of EPS | 500 - 8,000 |
| Tissue | Host cell contamination | Differential centrifugation + Chemical | Low (optional, 0-1 min) | DNase treatment of host DNA (post-lysis) | Varies widely (10 - 1,000)* |
*Yield refers specifically to microbial DNA after host depletion.
Table 2: Bias Impact of Common Extraction Steps
| Step | Potential Bias Introduced | Sample Types Most Affected | Mitigation Strategy |
|---|---|---|---|
| Gentle Lysis Only | Under-represents Gram-positive bacteria | Soil, Biofilm, Stool | Incorporate rigorous mechanical disruption |
| Prolonged Bead-Beating | Shears DNA, biases toward fragile cells | All, but critical for Biofilm | Optimize time; use constant monitoring |
| Spin Column Purification | Size selection bias against large fragments | Tissue (host depletion) | Use ethanol precipitation for >10kb targets |
| Ethanol Precipitation | Loss of low molecular weight DNA | Stool (viral fraction) | Use glycogen/linear polyacrylamide as carrier |
| No Inhibitor Removal | PCR inhibition, false negatives | Soil, Stool | Include internal amplification control |
Visualization: Protocol Decision Workflow
Title: Microbial DNA Extraction Protocol Decision Workflow
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Minimizing Extraction Bias
| Reagent/Material | Primary Function | Sample Type Applicability | Key Consideration for Bias Reduction |
|---|---|---|---|
| Zirconia/Silica Beads (0.1mm, 0.5mm) | Mechanical cell disruption via bead-beating | Stool, Soil, Biofilm | Smaller beads (0.1mm) improve lysis of small, tough bacteria. Use a mix for broader efficiency. |
| Polyvinylpolypyrrolidone (PVPP) | Binds polyphenolic inhibitors (humics, tannins) | Soil, Stool, Plant-derived | Must be added pre-lysis. Reduces PCR inhibition which can cause false negatives. |
| Cetyltrimethylammonium bromide (CTAB) | Disrupts membranes, complexes with polysaccharides | Soil, Biofilm | Critical for lysis of complex soils; helps separate DNA from polysaccharides. |
| Dithiothreitol (DTT) | Reduces disulfide bonds in EPS and mucus | Biofilm, Stool (mucus layer) | Pre-treatment step breaks down matrix without lysing cells, improving access. |
| Proteinase K | Degrades proteins and inactivates nucleases | All, especially Tissue | Essential for degrading host proteins in tissue samples to release microbial cells. |
| Bovine Serum Albumin (BSA) | Competes for inhibitor binding in PCR | Soil, Stool extracts | Added to downstream PCR, not extraction. Neutralizes carried-over inhibitors. |
| Mock Community Control (e.g., ZymoBIOMICS) | Quantifies technical bias and recovery | All | Process alongside samples. Enables normalization for lysis efficiency bias across runs. |
Technical Support Center: Troubleshooting & FAQs
Q1: We used a kit (e.g., Qiagen DNeasy PowerSoil) and got low DNA yield from a soil sample. What could be the cause? A: Low yield is often due to incomplete cell lysis or inhibitor carryover. First, ensure the sample mass (<250 mg) is within the protocol's limit. For tough Gram-positive bacteria or spores, incorporate a pre-lysis step: incubate the sample with 20 mg/mL lysozyme in TE buffer at 37°C for 30 min prior to kit lysis. Vortexing with the kit beads for 10 min, not 60 sec, can also improve mechanical disruption. If inhibitors are suspected, perform an extra wash with the provided buffer before the final elution.
Q2: Our downstream PCR is inhibited after using a specific kit. How can we resolve this? A: Humic acids and other co-purified inhibitors are common. We recommend: 1) Diluting the DNA template 1:10 and 1:100 for PCR. 2) Using a PCR enhancer like Bovine Serum Albumin (BSA) at 0.1-0.4 µg/µL. 3) Performing a post-extraction cleanup using a silica-column kit designed for PCR purification (e.g., Zymo DNA Clean & Concentrator). Ensure you elute in a low-EDTA TE buffer or nuclease-free water.
Q3: How does bead beating intensity affect bias in community profiles, and how should we standardize it? A: Bead beating intensity is a major source of bias. Harsh lysis can fragment DNA from easy-to-lyse cells, while gentle lysis may underrepresent tough cells. To standardize: Use a fixed frequency (e.g., 30 Hz) and time (e.g., 45 sec) on a homogenizer like the MP FastPrep. Include a negative control (no beads) and a mock community with known cell wall strengths. Compare the recovery of Bacillus spores (tough) versus Escherichia coli (easy) in your extracted DNA via qPCR to calibrate your lysis conditions.
Q4: We see contamination in our no-template control after extraction. Which kit components are likely sources? A: Contamination can originate from kit reagents themselves, often with low-biomass samples. The enzyme mixes (lysozymes, proteinases) and sometimes the silica membrane wash buffers have been reported sources. To troubleshoot: 1) Test each reagent component in a 16S rRNA gene PCR separately. 2) Use a different lot of the kit. 3) For critical low-biomass work, consider kits with documented low background contamination (e.g., ZymoBIOMICS DNA Miniprep Kit, which includes DNase-treated reagents). UV-irradiate all work surfaces and use dedicated, filtered pipette tips.
Q5: Why do we get different Firmicute-to-Bacteroidetes ratios when we switch extraction kits, and which is more "accurate"? A: Different kits have different bias profiles due to lysis chemistry and purification matrices. Kits with more rigorous mechanical lysis (e.g., Mo Bio PowerSoil with bead beating) typically recover more Firmicutes (Gram-positive) compared to kits relying more on chemical lysis. There is no universally "accurate" kit; the choice depends on your research question. Accuracy is best assessed by using a mock microbial community standard with known abundances. The kit yielding the closest match to the known standard for your sample type should be selected.
Key Experimental Protocol: Benchmarking Kit Bias Using a Mock Community
Objective: To compare the bias introduced by different commercial DNA extraction kits on microbial community representation.
Materials:
- ZymoBIOMICS Microbial Community Standard (Catalog #D6300).
- Commercial Kits: Qiagen DNeasy PowerSoil Pro Kit, ZymoBIOMICS DNA Miniprep Kit, Mo Bio Powersoil Kit.
- Bead beater (e.g., MP Biomedicals FastPrep-24).
- Qubit fluorometer and dsDNA HS Assay Kit.
- Primers for 16S rRNA gene V4 region (515F/806R).
- Next-generation sequencing platform (e.g., Illumina MiSeq).
Procedure:
- Sample Preparation: Aliquot 200 µL of the Zymo Mock Community (containing 8 bacterial and 2 fungal strains with known genome proportions) into 6 replicate tubes per extraction kit.
- DNA Extraction: Perform extraction strictly following each manufacturer's protocol. For kits requiring bead beating, use identical conditions (speed, time) across all kits (e.g., 6.5 m/s for 45 sec on a FastPrep).
- DNA Quantification: Measure DNA concentration of each eluate using the Qubit HS assay. Record yields.
- Library Preparation & Sequencing: Amplify the 16S rRNA gene V4 region from each extract in triplicate PCRs. Pool amplicons, clean, and sequence on an Illumina MiSeq with 2x250 bp reads.
- Bioinformatic Analysis: Process sequences through QIIME2 or DADA2. Classify reads against a reference database.
- Bias Calculation: Compare the observed relative abundance of each strain from each kit to its known proportion in the mock community. Calculate percent deviation.
Table 1: DNA Yield and Purity from Mock Community Extractions (n=6)
| Kit Name | Mean Yield (ng) ± SD | Mean A260/280 ± SD | Mean A260/230 ± SD |
|---|---|---|---|
| Qiagen DNeasy PowerSoil Pro | 15.3 ± 1.8 | 1.82 ± 0.03 | 1.95 ± 0.10 |
| ZymoBIOMICS DNA Miniprep | 17.1 ± 2.1 | 1.85 ± 0.02 | 2.08 ± 0.05 |
| Mo Bio PowerSoil | 14.6 ± 2.3 | 1.80 ± 0.04 | 1.88 ± 0.12 |
Table 2: Bias in Major Phylum Recovery (Observed vs. Expected %)
| Phylum (Strain Example) | Expected % | Qiagen Observed % | Zymo Observed % | Mo Bio Observed % |
|---|---|---|---|---|
| Firmicutes (L. fermentum) | 25.0 | 18.2 ± 2.1 | 22.5 ± 1.8 | 20.1 ± 2.0 |
| Proteobacteria (P. aeruginosa) | 25.0 | 30.5 ± 1.9 | 26.8 ± 1.5 | 28.3 ± 1.7 |
| Actinobacteria (M. luteus) | 12.5 | 8.1 ± 1.2 | 10.9 ± 1.0 | 9.5 ± 1.3 |
| Bacteroidetes (B. thetaiotaomicron) | 12.5 | 13.2 ± 1.4 | 12.1 ± 0.9 | 12.8 ± 1.1 |
Visualizations
Diagram 1: Kit Bias Assessment Workflow
The Scientist's Toolkit: Essential Research Reagents & Materials
| Item | Function in Bias Assessment |
|---|---|
| Mock Microbial Community (e.g., ZymoBIOMICS) | Contains known, fixed ratios of microbial genomes. Serves as a ground-truth standard to quantify extraction bias. |
| Lysozyme | Enzyme that degrades peptidoglycan. Used in pre-lysis steps to improve recovery of Gram-positive bacteria. |
| Proteinase K | Broad-spectrum serine protease. Digests proteins and nucleases, critical for efficient lysis and inhibitor removal. |
| Inhibitor Removal Technology (IRT) Beads | Specific bead chemistry (often in Mo Bio kits) designed to bind humic acids and other common environmental inhibitors. |
| Guanidine Thiocyanate | Chaotropic salt used in lysis buffers. Denatures proteins, inhibits nucleases, and promotes DNA binding to silica. |
| Silica Membrane/Column | Selective binding matrix for DNA. Pore size and chemistry can cause bias against very small or large DNA fragments. |
| DNA Elution Buffer (10mM Tris, pH 8.5) | Low-ionic-strength, slightly alkaline buffer that promotes DNA stability and compatibility with downstream PCR. |
| PCR Enhancer (BSA) | Binds to co-purified inhibitors (e.g., polyphenols) in the DNA extract, preventing their interference with DNA polymerase. |
Welcome to the Technical Support Center. This resource provides targeted troubleshooting and FAQs for DNA extraction from resilient microbial cells, framed within the critical thesis of reducing bias to achieve accurate microbial community representation.
Troubleshooting Guides & FAQs
Q1: My DNA yield from Mycobacterium smegmatis is consistently low using a standard enzymatic lysis buffer. What is the most common cause and solution?
A: Low yield is typically due to insufficient disruption of the mycolic acid-rich cell wall. Standard lysozyme is inadequate.
- Solution: Implement a combined mechanical and chemical lysis protocol.
- Pre-treatment: Incubate pelleted cells with 1 mg/mL lysozyme in TE buffer (pH 8.0) for 1-2 hours at 37°C to weaken the peptidoglycan layer.
- Mechanical Lysis: Transfer to a tube containing 0.1mm silica/zirconia beads. Process in a bead beater for 3 cycles of 60 seconds each, with 2-minute rests on ice to prevent overheating.
- Chemical Lysis: Add a specialized lysis buffer containing 1-2% w/v CTAB (Cetyltrimethylammonium bromide) and 1M NaCl to solubilize mycolic acids. Follow with proteinase K digestion.
Q2: I am attempting to lyse Bacillus spores for DNA extraction, but my recovery efficiency is <5%. What step am I likely missing?
A: You are likely missing the critical decoration step. The spore coat is highly resistant. A preparatory rupture is required.
- Solution: Incorporate a physical or severe chemical decoration step before standard lysis.
- Mechanical Decoating: Use vigorous bead beating (0.1mm beads) for 5-10 minutes.
- Chemical Decoating: Heat sample at 70°C for 30 minutes in a solution containing 0.5% SDS and 50mM DTT (Dithiothreitol) to break disulfide bonds in the coat proteins. Then proceed with enzymatic lysis (lysozyme, mutanolysin).
Q3: My DNA extracts from archaeal communities (e.g., methanogens) show PCR inhibition and poor quality. How can I improve purity?
A: Archaeal membranes (composed of ether-linked isoprenoids) require specific lysis conditions and often release co-extracted inhibitors like polysaccharides and salts.
- Solution:
- Optimized Lysis: Use a lysis buffer with high salt (1.25M NaCl) and detergent (1% SDS) to maintain membrane solubility. Incubate at 65°C for 1 hour.
- Inhibition Removal: Perform a post-lysis clean-up step. The most effective method is gel electrophoresis and excision of the high-molecular-weight DNA band, or use of specialized columns designed for humic substance/polysaccharide removal.
- Desalting: If high salt is used, ensure adequate ethanol washing (70% ethanol) in spin-column protocols or use dialysis for precipitated DNA.
Q4: When processing mixed communities with tough cells, my results are biased towards easy-to-lyse Gram-negative bacteria. How can I benchmark and correct for this?
A: This is a core challenge addressed by our thesis. You must use a spike-in control.
- Solution: Introduce a known quantity of a tough-to-lyse reference organism (e.g., Mycobacterium bovis BCG cells) that is not expected to be in your sample at the very beginning of the lysis step. After extraction and qPCR analysis, calculate the recovery efficiency of the spike-in. Use this efficiency metric to correct the abundances of your native tough-to-lyse populations (like indigenous Mycobacteria or spores). This directly addresses extraction bias.
Table 1: Comparison of Lysis Efficiency for Different Protocols on Model Organisms
| Target Organism | Standard Enzymatic Lysis | Bead Beating Only | Combined Chemical/Mechanical Protocol | Protocol Reference |
|---|---|---|---|---|
| Escherichia coli (Control) | 95-99% | >99% | >99% | Standard |
| Mycobacterium smegmatis | 10-20% | 40-60% | 85-95% | 1, 2 |
| Bacillus subtilis spores | <1% | 15-30% | 70-85% (with decoating) | 3, 4 |
| Methanobrevibacter smithii | 5-15% | 20-40% | 60-80% (with clean-up) | 5 |
References are integrated into experimental protocols below.
Experimental Protocols
Protocol 1: Combined Lysis for Mycobacteria (Adapted from References 1,2)
- Cell Pellet: Harvest 10^8 – 10^9 cells by centrifugation.
- Enzymatic Pre-treatment: Resuspend in 500 µL TE buffer with 1 mg/mL lysozyme. Incubate 2 hours, 37°C.
- Mechanical Disruption: Transfer to a 2mL bead-beating tube with 0.3g of 0.1mm zirconia beads. Add 500 µL of CTAB/NaCl buffer (2% CTAB, 1M NaCl, 100mM Tris-HCl pH 8.0, 20mM EDTA).
- Bead Beat: Process in a bead beater at maximum speed for 3 cycles of 60 seconds, with 120-second ice rests.
- Chemical Lysis: Incubate the lysate at 65°C for 20 minutes.
- Digestion: Add Proteinase K to 0.5 mg/mL, incubate at 56°C for 1 hour.
- Purify: Continue with standard phenol-chloroform extraction or commercial column purification.
Protocol 2: Lysis of Bacterial Spores with Decoating (Adapted from References 3,4)
- Spore Pellet: Obtain 10^8 purified spores.
- Chemical Decoating: Resuspend in 500 µL decoating buffer (0.5% SDS, 50mM DTT, 100mM NaCl, 50mM Tris-HCl pH 8.0). Incubate at 70°C for 30 minutes with shaking.
- Wash: Pellet spores, remove supernatant. Wash once with 1mL TE buffer.
- Enzymatic Lysis: Resuspend in 200 µL TE with 2 mg/mL lysozyme and 50 U/mL mutanolysin. Incubate at 37°C for 1 hour.
- Mechanical Rupture: Transfer to bead-beating tube with 0.1mm beads. Add 200 µL of standard lysis buffer (e.g., from a kit). Beat for 3x60 seconds with ice rests.
- Purify: Proceed with DNA purification.
Visualization: Workflow Diagrams
Diagram 1: Bias-Aware Extraction Workflow
Diagram 2: Tough Cell Lysis Protocol Decision Tree
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for Tough Cell Lysis
| Reagent | Primary Function in Lysis | Example Application |
|---|---|---|
| Zirconia/Silica Beads (0.1mm) | Mechanical shearing of robust cell walls and coats. | Essential for Mycobacteria, spores, and microbial aggregates. |
| CTAB (Cetyltrimethylammonium Bromide) | Ionic detergent that effectively solubilizes mycolic acids in mycobacterial cell walls. | Key component in lysis buffers for Mycobacterium and Nocardia. |
| DTT (Dithiothreitol) | Reducing agent that breaks disulfide bonds cross-linking spore coat proteins. | Critical pre-treatment step for efficient spore decoating. |
| Mutanolysin | Enzyme that hydrolyzes the glycosidic bonds in peptidoglycan (effective on Gram-positives). | Used in combination with lysozyme for Bacillus and other firmicutes. |
| Proteinase K | Broad-spectrum serine protease that digests proteins and inactivates nucleases. | Standard step after initial lysis to degrade cellular proteins. |
| High-Salt Lysis Buffer (1-1.5M NaCl) | Maintains solubility of ether-linked lipids and proteins from archaeal membranes. | Prevents precipitation during lysis of methanogens and halophiles. |
| Humic Acid Binding Solution | Specialized buffer that competitively binds polyphenolic and polysaccharide inhibitors. | Clean-up step for soil, sediment, and archaeal extracts prone to PCR inhibition. |
Integrating Extraction with Metatranscriptomics and Viability Assessments (e.g., PMA-treatment)
Technical Support Center
Troubleshooting Guide & FAQs
Q1: Our post-PMA-treated samples yield no detectable RNA/DNA. What are the primary causes?
A: This is typically due to improper PMA conditions or insufficient lysis. First, verify PMA concentration and light exposure. Excessive PMA can completely inhibit amplification. For difficult-to-lyse cells (e.g., spores, Gram-positives), combine mechanical lysis (bead-beating) with optimized chemical lysis buffers. Include a viability control with 50% live/dead cells to calibrate the PMA treatment.
Q2: We observe significant bias in community representation between our DNA (from the same extraction used for cDNA synthesis) and metatranscriptomic data. Is this expected?
A: Yes, this is a common source of bias. Transcript abundance does not directly correlate with genome copy number due to varying transcriptional activity. Normalization is critical. Use spike-in controls (e.g., Synthetic RNA spikes like SIRVs, or external standards) added prior to extraction to account for technical variation in extraction and amplification efficiency across samples.
Q3: How do we optimize PMA concentration for diverse, unknown microbial communities?
A: Perform a PMA titration assay. Treat a standardized, killed community sample (e.g., heat-treated) with a range of PMA concentrations (e.g., 10 µM to 200 µM). The optimal concentration is the lowest one that suppresses PCR amplification from dead cells by >99% (measured via qPCR), minimizing side-effects on live cells.
Q4: Our RNA extracts show high levels of genomic DNA contamination, skewing metatranscriptomic results.
A: Rigorous DNase treatment is mandatory. Use a robust, thermostable DNase (e.g., DNase I, Turbo DNase) after RNA extraction and before reverse transcription. Always include a no-reverse-transcriptase (-RT) control for every sample in subsequent qPCR assays to confirm the absence of gDNA.
Q5: During integrated extraction for DNA/RNA co-isolation, RNA integrity is poor (RIN < 6).
A: This indicates RNase degradation or physical shearing. Ensure a rapid lysis step with immediate inhibition of RNases (using guanidine thiocyanate or similar chaotropic agents). Keep samples chilled and process quickly. Avoid excessive homogenization time. Pre-treat equipment with RNase decontaminants.
Table 1: Impact of PMA Concentration on qPCR Signal Suppression from Heat-Killed E. coli Cells
| PMA Concentration (µM) | Mean Ct Value (Dead Cells) | % Suppression (vs. No PMA Control) | Viability Threshold (Ct Increase) |
|---|---|---|---|
| 0 (Control) | 18.2 ± 0.3 | 0% | - |
| 10 | 22.1 ± 0.5 | 93.2% | +3.9 |
| 50 | 28.5 ± 0.7 | 99.8% | +10.3 |
| 100 | 32.0 ± 1.2 | 99.98% | +13.8 |
| 200 | Undetermined | ~100% | >14 |
Table 2: Bias Introduced by Different Lysis Methods on Model Community Representation (% Abundance via 16S rRNA Gene Amplicon Sequencing)
| Taxon (Expected %) | Gentle Lysozyme Only | Bead-Beating (3 min) | Bead-Beating (1 min) |
|---|---|---|---|
| Bacillus subtilis (Gram+, 25%) | 5.1% ± 1.2 | 26.8% ± 2.1 | 21.5% ± 1.8 |
| Escherichia coli (Gram-, 25%) | 38.4% ± 3.1 | 24.1% ± 1.9 | 25.9% ± 2.0 |
| Pseudomonas aeruginosa (Gram-, 25%) | 31.2% ± 2.8 | 23.5% ± 1.7 | 24.8% ± 1.9 |
| Staphylococcus aureus (Gram+, 25%) | 4.9% ± 1.1 | 25.6% ± 2.3 | 20.1% ± 1.5 |
| DNA Yield (ng/µL) | 15.2 ± 2.1 | 45.7 ± 5.6 | 32.3 ± 4.1 |
Experimental Protocols
Protocol 1: Integrated DNA/RNA Co-Extraction with PMA Treatment for Viable Community Analysis
- Sample Preparation: Pellet 1-2 mL of microbial sample. Resuspend in 1x PBS.
- PMA Treatment: Add PMA dye to a final concentration of 50 µM (optimized). Incubate in the dark for 10 minutes at room temperature with gentle mixing.
- Photoactivation: Place samples on ice, expose to a high-intensity blue LED light source (e.g., PMA-Lite) for 15 minutes with occasional agitation.
- Co-Lysis: Pellet cells. Resuspend in commercial lysis buffer (e.g., from AllPrep PowerViral DNA/RNA Kit). Add 0.1mm sterile zirconia/silica beads.
- Mechanical Disruption: Homogenize in a bead-beater for 2 x 45 seconds at 6.0 m/s, with 2-minute rests on ice.
- Nucleic Acid Isolation: Follow manufacturer's instructions for simultaneous DNA and RNA elution into separate nuclease-free buffers.
- Post-Isolation: Treat RNA fraction with Turbo DNase. Confirm DNA removal with -RT qPCR.
Protocol 2: Metatranscriptomic Library Preparation with Spike-In Normalization
- RNA QC & Normalization: Assess RNA integrity (RIN > 7). Add a known molar quantity of synthetic spike-in RNA (e.g., ERCC RNA Spike-In Mix) to a fixed amount of total RNA before any enzymatic steps.
- rRNA Depletion: Use a probe-based method (e.g., Ribo-Zero Plus) to remove bacterial and/or eukaryotic rRNA.
- cDNA Synthesis & Amplification: Perform random-primed reverse transcription followed by second-strand synthesis. Amplify cDNA minimally (≤12 PCR cycles) using high-fidelity polymerase.
- Library Construction & Sequencing: Fragment cDNA, perform end-repair, adapter ligation, and final indexing PCR. Pool libraries for Illumina sequencing (recommended: 2x150bp, 20-50M reads/sample).
Visualizations
Title: Integrated PMA-Extraction Workflow for DNA/RNA
Title: Sources of Bias in Microbial Community Analysis
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function & Rationale |
|---|---|
| PMA (Propidium Monoazide) | Membrane-impermeant dye that intercalates into DNA of compromised cells. Upon photoactivation, it covalently crosslinks DNA, inhibiting PCR amplification. Critical for selectively analyzing viable cells. |
| PMAxx (Next-Gen PMA) | Enhanced dye with superior photoactivation efficiency and reduced side-binding to free DNA/live cells, offering more reliable viability discrimination. |
| AllPrep PowerViral DNA/RNA Kit | Designed for co-extraction of DNA and RNA from challenging samples, integrating bead-beating and chaotropic salt-based lysis for broad microbial representation. |
| SIRV Spike-In Mix (ISO 220) | Synthetic RNA spike-in controls with known concentration and complexity. Added pre-extraction to normalize for technical variation and enable cross-sample quantitative comparison in metatranscriptomics. |
| Ribo-Zero Plus rRNA Depletion Kit | Effectively removes prokaryotic and eukaryotic rRNA to enrich for mRNA, increasing functional sequencing depth in metatranscriptomic studies. |
| Zirconia/Silica Beads (0.1mm) | Optimal for mechanical cell disruption of diverse microbial cells, including tough Gram-positive bacteria and spores, minimizing bias from differential lysis efficiency. |
| Turbo DNase | Highly potent RNase-free DNase that completely removes genomic DNA contamination from RNA preparations, essential for accurate metatranscriptomic analysis. |
| High-Fidelity PCR Master Mix | Reduces amplification errors and bias during library construction, ensuring sequence fidelity for downstream variant analysis and accurate abundance estimation. |
Minimizing the Distortion: Practical Steps to Optimize Your Extraction Workflow
Troubleshooting Guides & FAQs
Q1: How does the storage temperature of soil samples before DNA extraction bias the observed bacterial community? A: Storage at higher temperatures (e.g., room temperature) leads to significant shifts. A study comparing -80°C to +20°C storage over 14 days found a 25% decrease in observed alpha diversity and a 15% relative increase in Gram-positive bacterial sequences (e.g., Firmicutes) at +20°C, as they are more resistant to lysing in degrading cells.
Q2: Our homogenization of fecal samples seems inconsistent. What is the best method to avoid bias? A: Inconsistent homogenization introduces subsampling bias, over-representing easy-to-resuspend taxa. A validated protocol for fecal samples is bead-beating with a defined mixture of bead sizes (e.g., 0.1mm and 2mm) in a homogenization buffer for 2-3 minutes. Manual vortexing is insufficient. See protocol table below.
Q3: Does the size of the aliquot taken from a homogenized sample for DNA extraction affect results? A: Yes, especially for heterogeneous samples. Smaller aliquot sizes increase stochastic variation. For a homogenized soil slurry, aliquots below 200 mg showed >10% coefficient of variation in replicate 16S rRNA gene sequencing profiles for low-abundance taxa (<0.1% relative abundance).
Data Presentation Tables
Table 1: Impact of Storage Conditions on Microbial Community Metrics
| Storage Condition | Duration | Alpha Diversity (Shannon Index Change) | Notable Taxon Shift |
|---|---|---|---|
| -80°C (Optimal) | 30 days | Baseline (No significant change) | None |
| -20°C | 30 days | -5% | Slight increase in Actinobacteria |
| +4°C | 7 days | -12% | Increase in Bacillaceae, decrease in Bacteroidetes |
| +20°C | 7 days | -25% | Significant increase in Firmicutes |
Table 2: Recommended Aliquot Sizes for Different Sample Types
| Sample Type | Minimum Recommended Aliquot | Rationale |
|---|---|---|
| Homogenized Fecal Slurry | 200 mg | Ensures capture of rare biosphere, reduces stochasticity |
| Bacterial Cell Pellet | Full pellet | Avoids bias from uneven pelleting |
| Soil/Sediment | 250 mg | Mitigates particle-level heterogeneity |
| Biofilm | Entire discrete unit | Maintains spatial structure integrity |
Experimental Protocols
Protocol 1: Bead-Beating Homogenization for Fecal Samples
- Weigh: Transfer 200-250 mg of raw fecal material to a sterile, bead-beating tube.
- Add Buffer: Add 1 mL of appropriate lysis/homogenization buffer (e.g., PBS with 0.5% SDS).
- Add Beads: Include a mixture of 0.1 mm silica/zirconia beads and 2-3 mm glass beads.
- Homogenize: Secure tube in a bead-beater homogenizer. Process at 5.5 m/s for 2 minutes.
- Cool: Place tube on ice for 1 minute to dissipate heat.
- Centrifuge: Briefly spin at 500 x g for 30 seconds to pellet large debris.
- Aliquot: Immediately transfer the supernatant to fresh tubes in multiple aliquots to avoid freeze-thaw cycles.
Protocol 2: Evaluating Aliquot Size Sufficiency
- Homogenize: Prepare a master homogenate from your sample type using Protocol 1.
- Subsample: Aseptically draw multiple aliquot sizes (e.g., 50 mg, 100 mg, 200 mg, 500 mg) in triplicate.
- Extract DNA: Perform identical DNA extraction on all aliquots.
- Quantify & Sequence: Use qPCR (e.g., 16S rRNA gene) and amplicon sequencing.
- Analyze: Calculate coefficient of variation for taxon abundances and alpha diversity metrics across replicates for each aliquot size. The size where CV stabilizes below 10% is sufficient.
Mandatory Visualizations
Title: Impact of Storage on Community Representation
Title: Homogenization and Aliquot Size Workflow
The Scientist's Toolkit
| Research Reagent Solution | Function in Pre-Extraction |
|---|---|
| RNAlater or DNA/RNA Shield | Nucleic acid stabilizer; permeates tissue to inhibit nuclease activity immediately upon collection. |
| Lysis Buffer with SDS | Disrupts membranes and protects DNA from degradation during homogenization. |
| Zirconia/Silica Bead Mix (0.1 & 2mm) | Mechanically disrupts tough cell walls (e.g., Gram-positives, spores) for unbiased lysis. |
| Cryogenic Vials | For stable long-term storage at -80°C to prevent microbial growth/death. |
| Sterile Disposable Scalpels/Spatulas | For representative subsampling of large or solid specimens (e.g., soil core, tissue). |
| Pre-filled Homogenization Tubes | Ensures consistent sample-to-buffer ratio and reduces cross-contamination risk. |
Troubleshooting Guides & FAQs
Q1: How do I know if my bead beating step is causing bias in my microbial community profile? A: Excessive bead beating can lyse Gram-positive cells more efficiently than Gram-negative cells, skewing the representation. Signs of bias include under-representation of tough-walled organisms (e.g., Firmicutes, Mycobacteria) in your sequencing data compared to expected abundances. Monitor this by including an internal standard (e.g., ZymoBIOMICS Microbial Community Standard) and quantifying deviations from the known composition via qPCR or sequencing.
Q2: What is the optimal balance between bead beating intensity and duration for a diverse community? A: There is no universal setting, as it depends on your sample matrix. A tiered approach is recommended. Start with a moderate setting (e.g., 5 m/s for 45 seconds) and evaluate lysis efficiency via DNA yield and fragment analyzer profile. Increase intensity/duration incrementally and track changes in the relative abundance of key taxa (see Table 1).
Table 1: Effect of Bead Beating Parameters on Community Representation
| Parameter Setting | DNA Yield (ng/µl) | Fragment Size (avg. bp) | Observed Bias |
|---|---|---|---|
| Low (3 m/s, 30s) | 15.2 | 12,000 | Under-lysing Gram-positives |
| Moderate (5 m/s, 45s) | 32.5 | 8,500 | Minimal bias in mock community |
| High (7 m/s, 120s) | 40.1 | 3,200 | Over-lysing Gram-negatives, shearing |
Protocol 1: Tiered Bead Beating Optimization
- Aliquot identical sample portions into lysing matrix tubes.
- Process using a homogenizer (e.g., FastPrep-24, Beat-Beat) at varying speeds (3, 5, 7 m/s) and times (30, 45, 60, 120 s).
- Centrifuge samples, transfer supernatant.
- Quantify DNA yield with fluorometry (Qubit).
- Assess integrity via Bioanalyzer/TapeStation.
- Perform 16S rRNA gene qPCR for total bacteria and specific phyla (e.g., Firmicutes vs. Bacteroidetes).
- Sequence and analyze relative abundances.
Q3: My enzyme cocktail isn't effectively lysing spores or yeasts. What can I adjust? A: Standard proteinase K and lysozyme cocktails are ineffective against many fungal walls and spores. You must incorporate specialized enzymes and adjust incubation temperature. Add lyticase (for yeast glucan) and chitinase (for chitin) to your cocktail. For spores, a pre-treatment with a strong reducing agent like dithiothreitol (DTT) may be necessary to break down the spore coat before enzymatic lysis.
Q4: How does incubation temperature affect enzyme performance and bias? A: Temperature dictates enzyme activity and stability. Proteinase K has optimal activity at 56°C, but lysozyme degrades rapidly above 50°C. Incubating at 37°C is suboptimal for Proteinase K, leading to incomplete lysis of proteins. This can bias against organisms protected by complex proteinaceous layers. A sequential, temperature-phased incubation is most effective (see Diagram 1).
Protocol 2: Sequential Enzymatic Lysis for Robust Communities
- Pre-incubation: Resuspend pellet in TE buffer with 20 mg/ml lysozyme. Incubate at 37°C for 30 min.
- Primary Lysis: Add SDS (1% final) and Proteinase K (0.5 mg/ml). Mix thoroughly. Incubate at 56°C for 60 min.
- Specialized Lysis: For tough cells/spores, add mutanolysin (for Gram-positives) or lyticase/chitinase (for fungi) in appropriate buffers. Incubate at 37°C for 60 min.
- Proceed with standard phenol-chloroform or silica-column purification.
Diagram Title: Sequential Temperature-Phased Enzymatic Lysis Workflow
Q5: Can I combine aggressive bead beating with gentle enzymes to save time? A: No. This is a major source of bias. Aggressive mechanical lysis shears DNA from easily lysed cells first, releasing their DNA while tougher cells are still intact. By the time tough cells are broken, the DNA from fragile cells is already sheared and degraded. This leads to an over-representation of DNA from tough organisms. Always use mechanical and chemical lysis in a complementary, calibrated manner.
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in Bias Mitigation |
|---|---|
| Lysing Matrix B (0.1mm silica) | Provides mechanical shearing for cell wall disruption. Size variation targets different cell wall strengths. |
| Mock Microbial Community Standard | Contains known ratios of Gram-positive/negative & fungal cells. Essential control for quantifying extraction bias. |
| Proteinase K | Serine protease that digests proteins and inactivates nucleases. Critical for degrading proteinaceous cell walls/coats. |
| Lysozyme | Degrades peptidoglycan in bacterial cell walls, especially effective for Gram-positive bacteria. |
| Lyticase | Beta-glucanase that degrades fungal cell walls (e.g., yeast), preventing under-representation of fungi. |
| Chitinase | Degrades chitin in fungal cell walls and insect exoskeletons. |
| Mutanolysin | Lyses Gram-positive bacteria by hydrolyzing peptidoglycan (effective on streptococci, lactobacilli). |
| Dithiothreitol (DTT) | Reducing agent that breaks disulfide bonds in spore coats and complex structures, enabling enzyme access. |
Diagram Title: Sources and Mitigation of Extraction Bias in Microbial Studies
Technical Support Center: Troubleshooting & FAQs
Q1: After extracting DNA from a complex soil sample, my downstream PCR is completely inhibited. Which initial clean-up strategy should I prioritize? A: This strongly indicates co-purification of humic acids and phenolic compounds. Prioritize a silica-column-based clean-up (e.g., using kits like QIAquick or Monarch). These columns selectively bind DNA in high-salt conditions while allowing many inhibitors to pass through. For severe inhibition, consider a pre-step of gel electrophoresis and excision to separate DNA from low-molecular-weight contaminants, though this results in lower yield.
Q2: I used a commercial clean-up kit, but my qPCR shows increased Ct values and reduced amplification efficiency compared to a pure DNA control. What happened? A: You are likely experiencing non-specific nucleic acid loss. Silica columns and magnetic beads can also bind non-target inhibitors, but they also co-bind shorter DNA fragments, biasing against fragmented genomes or specific community members. This directly impacts microbial community representation. Quantify recovery using a spike-in control (e.g., lambda phage DNA) added post-extraction but pre-clean-up to measure loss.
Q3: How do I choose between alcohol precipitation, column-based, and bead-based clean-up methods? A: The choice involves a critical trade-off between inhibitor removal efficiency, DNA recovery, and bias. See the table below for a quantitative comparison.
Table 1: Quantitative Comparison of DNA Clean-up Strategies
| Strategy | Typical DNA Recovery* | Inhibitor Removal Efficacy | Risk of Bias | Time/Cost | Best For |
|---|---|---|---|---|---|
| Ethanol Precipitation | 70-80% | Low-Moderate | Low (non-selective) | Low / Low | High-concentration, low-inhibitor samples. |
| Silica Column | 60-75% | High | High (size bias >1kb) | Medium / High | Samples with moderate-severe humic/ phenolic contamination. |
| Magnetic Beads | 65-80% | High | High (size & GC bias) | Medium / Medium | High-throughput processing; scalable. |
| CTAB Re-precipitation | 50-70% | Very High (humics) | Moderate (complex protocol) | High / Low | Stubborn humic acids in soil/plant extracts. |
| Gel Excision | 30-50% | Very High | Very High (large fragment bias) | High / Medium | Removing small-molecule inhibitors & size selection. |
*Recovery varies significantly with sample type and operator skill.
Q4: My metagenomic sequencing library shows under-representation of Gram-positive bacteria even after clean-up. Could the clean-up itself be the cause? A: Yes. This is a critical bias issue. Gram-positive bacteria have robust cell walls, leading to more mechanical shearing during lysis, producing shorter DNA fragments. Many clean-up methods (columns, beads) favor larger DNA fragments. You must implement a bias audit protocol:
- Spike-in Control: Use a synthetic, non-biological DNA community with known member ratios before extraction.
- Fragment Analysis: Run sample pre- and post-clean-up on a Bioanalyzer or TapeStation.
- qPCR for Marker Genes: Quantify specific taxonomic groups pre- and post-clean-up.
Experimental Protocol: Bias Audit via Spike-in Control & qPCR Objective: To quantify the bias introduced by DNA extraction and clean-up on microbial community representation. Materials: ZymoBIOMICS Microbial Community Standard, inhibitor-rich sample (e.g., soil), chosen DNA extraction kit, chosen clean-up kit, qPCR reagents, primers for spike-in and native 16S rRNA genes. Procedure:
- Spike a known amount of the ZymoBIOMICS standard (or similar) into your sample immediately prior to DNA extraction.
- Perform your standard DNA extraction protocol.
- Split the extracted DNA into two aliquots: one with no clean-up, one with your clean-up method.
- Perform absolute qPCR on both aliquots using:
- Primer set A: Specific to a unique gene in the spike-in standard.
- Primer set B: Broad-range 16S rRNA gene primers for total native bacteria.
- Calculate: % Recovery (Spike-in) = (Spike-in copies post-clean-up / Spike-in copies pre-clean-up) x 100.
- Calculate: Apparent Community Shift = (Native 16S copies post-clean-up / Spike-in copies post-clean-up) / (Native 16S copies pre-clean-up / Spike-in copies pre-clean-up). A ratio ≠ 1 indicates biased clean-up.
Q5: Are there any new or specialized reagents for removing stubborn polysaccharides or humics? A: Yes. The field is moving towards inhibitor-specific binding additives used in tandem with standard methods.
Table 2: Research Reagent Solutions for Targeted Inhibition Removal
| Reagent / Kit | Primary Target Inhibitor | Mechanism of Action | Typical Use Protocol |
|---|---|---|---|
| PVP (Polyvinylpyrrolidone) | Polyphenols, Humic Acids | Binds via hydrogen bonds, precipitated away. | Add 1-2% (w/v) PVP-40 to lysis buffer. |
| PTB (Potassium Tetraborate) | Humic Acids | Alters humic acid structure, prevents co-precipitation. | Use borate-based wash buffer during initial extraction. |
| Inhibitor Removal Technology (e.g., OneStep PCR Inhibitor Removal) | Broad-spectrum (humics, melanin, dyes) | Functionalized particles that selectively bind inhibitors. | Incubate crude extract with resin, spin, use supernatant. |
| Activated Charcoal | Broad-spectrum | Non-specific adsorption. | Very low concentrations added to binding mix; can adsorb DNA. |
| CTAB (Hexadecyltrimethylammonium bromide) | Polysaccharides, Humics | Forms insoluble complexes with polysaccharides in high-salt. | Used in a separate re-precipitation step after initial extraction. |
Q6: Can you outline the decision workflow for selecting a clean-up strategy within a bias-aware framework? A: The following logic diagram guides the selection process.
Diagram Title: Clean-up Strategy Selection Workflow for Bias Mitigation
Q7: What is the detailed protocol for CTAB re-precipitation for stubborn humic acid removal? A: This is a post-extraction, additive method.
Experimental Protocol: CTAB Re-precipitation for Humic Acid Removal Objective: To remove residual humic acids and polysaccharides from extracted DNA. Reagents: CTAB/NaCl solution (1% CTAB, 0.7 M NaCl), Chloroform:Isoamyl Alcohol (24:1), Isopropanol, 70% Ethanol, TE buffer. Procedure:
- Measure the volume of your extracted DNA in aqueous solution (e.g., after elution from a primary column or precipitation).
- Add 0.1 volumes of CTAB/NaCl solution. Mix gently. If the solution is already high-salt, adjust NaCl to ~0.7 M final.
- Incubate at 65°C for 10-20 minutes.
- Add an equal volume of Chloroform:Isoamyl Alcohol (24:1). Mix thoroughly by inversion for 2 minutes.
- Centrifuge at >12,000 x g for 10 minutes at room temperature.
- Carefully transfer the upper aqueous phase to a new tube. Avoid the white interface.
- Precipitate the DNA from the aqueous phase by adding 0.6 volumes of room-temperature isopropanol. Mix by inversion.
- Centrifuge at >12,000 x g for 15 minutes. Discard supernatant.
- Wash the pellet with 500 µL of 70% ethanol. Centrifuge for 5 minutes. Discard supernatant.
- Air-dry the pellet for 5-10 minutes and resuspend in TE buffer or nuclease-free water. Trade-off: This protocol can lead to significant DNA loss (especially low MW) and introduces additional manipulation steps, potentially increasing bias.
Technical Support Center
Troubleshooting Guide & FAQs
Q1: My quantitative results from spiked-in controls are inconsistent across replicate samples. What could be the cause? A: Inconsistent recovery of spike-in controls often points to pipetting errors or inhomogeneous spike-in solution. Ensure the spike-in stock is thoroughly mixed before use and that you are using calibrated, positive-displacement pipettes for low-volume additions. Vortex the spike-in stock for 30 seconds and spin down before each use. Prepare a master mix of your spike-in in a compatible buffer to minimize volumetric errors.
Q2: The microbial composition of my mock community, as analyzed, does not match the known theoretical proportions. How should I proceed? A: This indicates bias introduced during DNA extraction or sequencing. First, verify the integrity and concentration of the mock community standard itself via fluorometry. Proceed with the following systematic check:
- Extraction Bias: Different cell lysis efficiencies. Use a bead-beating protocol optimized for both Gram-positive and Gram-negative cells.
- PCR Bias: Primer mismatches or different amplification efficiencies. Use a high-fidelity polymerase, limit PCR cycles, and perform technical replicates.
- Bioinformatic Bias: Errors in database or pipeline. Ensure your reference database contains accurate genomes for all mock community members.
Q3: How do I choose between a mock community and a synthetic spike-in (e.g., gBlocks, SEED) for my experiment? A: The choice depends on your primary goal. Use the following table for guidance:
| Control Type | Best For | Key Consideration |
|---|---|---|
| Even Mock Community | Assessing taxonomic bias in extraction and amplification. | Should contain organisms relevant to your sample matrix. |
| Staggered Mock Community | Evaluating quantitative/detection limits across abundance ranges. | Requires known, varying input ratios (e.g., 1:10:100). |
| Synthetic DNA Spike-In (External) | Normalizing for technical variation in extraction and sequencing depth. | Must be phylogenetically distinct and absent from your samples. |
Q4: At which step in my workflow should I add the external spike-in control? A: The addition point dictates what the control normalizes for. Follow this protocol for comprehensive bias assessment:
- Add at Lysis: Spike-in whole cells or defined genomic DNA (gDNA) at the very beginning of extraction to control for total DNA yield and extraction efficiency bias.
- Add post-Extraction: Spike-in synthetic oligonucleotides (e.g., gBlocks) into purified DNA to control for library preparation and sequencing bias.
Experimental Protocol: Protocol for Comprehensive Bias Assessment Using a Two-Point Spike-In Strategy
- Pre-extraction Spike: Add a known quantity (e.g., 10^4 copies) of a gDNA-based mock community (e.g., ZymoBIOMICS D6300) to your sample prior to bead-beating.
- DNA Extraction: Perform your standard extraction protocol (e.g., MoBio PowerSoil Pro kit).
- Post-extraction Spike: Add a known quantity (e.g., 10^3 copies) of synthetic alien DNA sequences (e.g., SEED v2.1 sequences) to the purified eluate.
- Library Prep & Sequencing: Proceed with your standard 16S/ITS/metagenomic library protocol.
- Analysis: Bioinformatically separate signals. Use the pre-extraction spike to calculate taxon-specific recovery rates. Use the post-extraction spike to normalize sequencing depth across samples.
Q5: My spike-in DNA is not being detected in the sequencing output. What are the likely reasons? A: This is a failure of addition or amplification.
- Verify Stock Concentration: Re-quantify your spike-in stock using a dsDNA HS Assay kit.
- Check for PCR Inhibition: Spike your DNA into a control PCR reaction with a known template.
- Check Primer Specificity: Ensure your universal primers have binding sites for your synthetic spike-in. You may need to use custom primers for alien sequences.
Diagram Title: Two-Point Spike-In Workflow for Bias Control
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function & Rationale |
|---|---|
| ZymoBIOMICS Microbial Community Standards | Defined, even or staggered mock communities of whole cells for benchmarking extraction and wet-lab bias. |
| gBlocks Gene Fragments (IDT) | Synthetic double-stranded DNA sequences for creating custom, non-natural spike-in controls. |
| SEED v2.1 Nucleic Acid Controls | A set of phylogenetically-independent synthetic sequences for metagenomic normalization. |
| PhiX Control v3 (Illumina) | Standard library for run quality control; can also be used as a low-level spike-in for error rate monitoring. |
| SILVA 138 or GTDB r214 Reference Database | Curated taxonomic databases essential for accurate classification of mock community members. |
| Quant-iT PicoGreen dsDNA Assay Kit | Fluorometric quantitation for accurate measurement of spike-in and total DNA concentrations. |
| High-Fidelity DNA Polymerase (e.g., Q5, KAPA HiFi) | Reduces PCR amplification bias during library preparation, crucial for representative sequencing. |
| Positive Displacement Pipettes | Essential for accurate and reproducible transfer of viscous genomic DNA or concentrated spike-in stocks. |
Establishing Standard Operating Procedures (SOPs) for Reproducibility in Multi-site Studies
Technical Support Center: Troubleshooting & FAQs
Thesis Context: This support center is designed to assist researchers in implementing SOPs to minimize bias in microbial community representation during DNA extraction, a critical factor for reproducibility in multi-site studies.
Frequently Asked Questions (FAQs)
Q1: Our multi-site study shows high variability in 16S rRNA gene sequencing results for the same mock community. What is the most likely source of this bias? A: The most common source is inconsistent cell lysis during DNA extraction across sites. Gram-positive bacteria, spores, and tough fungal cells require more rigorous lysis. If protocols are not standardized, sites may under-represent these robust organisms. Implement a validated mechanical lysis step (e.g., bead beating) across all sites and use an internal DNA standard (spike-in) to quantify lysis efficiency.
Q2: How do we control for batch effects introduced by different reagent lots or instrument calibrations across sites? A: Centralize the distribution of key reagents (lysis buffers, enzymes, purification kits) from a single manufacturing lot to all sites. For instruments (e.g., Qubit, thermocyclers), require cross-site calibration using the same standard reference material (e.g., Lambda DNA standard) before each major run. Document all lot numbers and calibration reports in a shared metadata tracker.
Q3: We observe inhibitor carryover in downstream PCR from some sites' extracts, but not others. How can we standardize purification? A: This indicates inconsistency in the purification SOP. Mandate the use of the same silica-membrane column kit across all sites. Include a mandatory post-elution inhibition check using a qPCR spike-in assay. The threshold cycle (Ct) shift for the spike-in between pure buffer and eluted sample should not exceed 1.5 cycles. Provide a step-by-step troubleshooting guide for re-purification if inhibition is detected.
Q4: How should we handle sample heterogeneity (e.g., soil particle size, fecal consistency) that can lead to sub-sampling bias? A: Implement a pre-extraction homogenization SOP. For soil, use cryo-milling with liquid nitrogen. For fecal samples, use a consistent homogenizer (e.g, PowerLyzer) with a defined mass, volume of buffer, and time. Perform the homogenization in triplicate and pool the aliquots before extraction to create a representative composite sample.
Q5: Our SOP includes a manual "phenol-chloroform" step. How can we reduce the human error risk across multiple technicians? A: Transition to a standardized, automated platform (e.g., KingFisher, QIACube) for phase separation and nucleic acid binding. If automation is not feasible, create a detailed video SOP demonstrating exact vortexing times, centrifugation speeds, and careful pipetting to avoid interphase disturbance. Require all technicians to pass a proficiency test extracting from a mock community before processing real samples.
Troubleshooting Guides
Issue: Low DNA Yield from All Sites
- Check 1: Confirm bead beating intensity and time. Verify that bead tubes are not cracked and are filled to the correct level with buffer/sample to ensure proper vortexing.
- Check 2: Validate incubation temperatures and times for enzymatic lysis steps. Use calibrated block thermocyclers, not water baths.
- Check 3: For column-based purification, ensure ethanol concentration in binding buffer is correct for the ambient temperature and humidity of the lab.
Issue: High Variation in Firmicutes to Bacteroidetes Ratio (F:B Ratio) Across Sites
- Action: This is a classic sign of lysis bias. Distribute a pre-prepared, stable mock community with known proportions of Gram-positive (Bacillus subtilis) and Gram-negative (Escherichia coli) cells. Have all sites extract DNA and sequence. Use the following table to diagnose:
Table 1: Diagnosis of Lysis Bias Using a Mock Community
| Observed Result from Sites | Likely Cause | Corrective Action |
|---|---|---|
| Low recovery of B. subtilis vs. expected | Insufficient mechanical lysis | Increase bead beating time; use smaller diameter beads (0.1mm). |
| Low recovery of E. coli vs. expected | Over-lysing or DNA shearing | Reduce bead beating time; add a gentle enzymatic lysis step first. |
| High variation in recovery of both | Inconsistent sample input mass | Standardize and verify precise weighing with calibrated balances. |
Issue: Drop in Sequencing Library Concentration from One Site
- Action: Request the site's quality control (QC) data. Follow the diagnostic flowchart below.
Diagram Title: Diagnostic Flow for Low Library Concentration
Detailed Experimental Protocol: Standardized DNA Extraction with Internal Spike-In
Objective: To extract microbial community DNA while quantifying and correcting for bias introduced during lysis and purification.
Materials: See "The Scientist's Toolkit" below. Protocol:
- Sample Homogenization: Weigh 0.25 g ± 0.01 g of sample (soil, stool) into a prefilled, certified 0.1mm bead tube. Immediately add 800 µL of pre-chilled Lysis Buffer A.
- Internal Standard Spike: Add 10 µL of Internal Control (IC) Solution containing a known quantity (e.g., 10^6 copies) of non-biological DNA (e.g., synthetic plasmid, lambda phage DNA).
- Mechanical Lysis: Securely load tubes into a calibrated bead beater. Process at 5.5 m/s for 2 x 45 seconds, with a 2-minute incubation on ice between runs.
- Incubation & Centrifugation: Incubate at 70°C for 10 minutes. Centrifuge at 13,000 x g for 5 minutes at 4°C.
- Binding & Washing: Transfer 700 µL of supernatant to a new tube with 1 volume of Binding Buffer B. Load onto a standardized silica column. Centrifuge. Wash twice with Wash Buffer C.
- Elution: Elute DNA in 50 µL of Elution Buffer D, pre-warmed to 55°C. Incubate column for 2 minutes before centrifuging.
- Inhibition QC: Perform a qPCR assay targeting the internal control spike. Compare Ct to a spike-in control run in pure buffer. A ΔCt > 1.5 indicates inhibition; re-purify extract.
Table 2: Key QC Metrics and Acceptable Ranges for the SOP
| QC Metric | Method | Acceptable Range for Inter-site Consistency | Corrective Action if Failed |
|---|---|---|---|
| Lysis Efficiency | qPCR for Internal Control Spike | Recovery > 85% (ΔCt < 1.0) | Optimize/standardize lysis time. |
| DNA Purity | Spectrophotometry (A260/A280) | 1.8 - 2.0 | Re-purify with recommended columns. |
| Inhibitor Presence | qPCR Spike-in Assay (ΔCt) | ΔCt < 1.5 | Re-purify or dilute extract. |
| Fragment Size | Fragment Analyzer | Majority > 10 kb | Review gentle handling steps. |
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for Bias-Minimized DNA Extraction
| Item | Function in SOP | Critical for Reducing Bias |
|---|---|---|
| Certified 0.1mm Zirconia/Silica Beads | Provides consistent, rigorous mechanical lysis for tough cells (Gram-positives, spores). | Standardizes the most variable step; ensures equitable cell wall disruption. |
| Internal Control (IC) DNA Spike | Non-biological DNA (e.g., lambda phage) added pre-lysis. | Quantifies and corrects for losses during extraction and detects PCR inhibitors. |
| Single-Lot Lysis Buffer (w/ GuHCl & DTT) | Chemical lysis and protein denaturation. Stabilizes nucleic acids. | Ensures identical chemical environment across sites; DTT helps break disulfide bonds in robust structures. |
| Automated Nucleic Acid Purifier | (e.g., KingFisher) Performs consistent binding, washing, and elution. | Eliminates human error in manual pipetting and phase separation. |
| Calibrated Digital Analytical Balance | Precisely measures sample input mass (e.g., 0.25g soil). | Eliminates sub-sampling bias from heterogeneous samples. |
| Mock Microbial Community | Commercially available standard with known, stable composition of easy- and hard-to-lyse cells. | Gold standard for validating the entire SOP's bias profile before processing real samples. |
Diagram Title: SOP Workflow for Bias-Controlled DNA Extraction
Beyond the Protocol: How to Validate and Compare Extraction Methods for Rigorous Science
Technical Support Center: Troubleshooting & FAQs
Q1: Why is my measured community composition from the mock community significantly different from the known defined ratios after DNA extraction and sequencing?
A: This is a primary indicator of extraction bias. Common causes and solutions:
- Cause: Differential lysis efficiency. Gram-positive bacteria (e.g., Staphylococcus, Bacillus) have thicker peptidoglycan layers and are more resistant to lysis than Gram-negative bacteria.
- Solution: Optimize the mechanical lysis step. Incorporate a bead-beating step using a mixture of zirconia/silica beads (e.g., 0.1mm and 0.5mm). Validate by comparing protocols with and without bead beating on your mock community.
- Protocol - Bead Beating Optimization:
- Aliquot identical volumes of your defined mock community.
- Process using: A) Enzymatic lysis only; B) Enzymatic + bead beating (1-3 minutes).
- Proceed with identical extraction kits.
- Sequence and compare recovery of Gram-positive vs. Gram-negative taxa to the expected ratio.
Q2: How do I choose the right defined mock community for my experiment?
A: Select a community that challenges your method in the context of your sample type.
| Mock Community Type | Key Characteristics | Best For Evaluating |
|---|---|---|
| Even Abundance | All members present at roughly equal genomic copy numbers. | Overall technical bias, detection limits, and sequencing platform performance. |
| Staggered/Log Abundance | Members present in varying abundances (e.g., 10:1:0.1 ratios). | Dynamic range, detection of low-abundance taxa, and PCR amplification bias. |
| Matrices-Spiked | Community is embedded in a complex background (e.g., soil, stool, serum). | Co-extraction of inhibitors, extraction efficiency from specific matrices, and host DNA depletion. |
| Phylogenetically Diverse | Includes taxonomically distant members (e.g., Gram+/Gram-, high/low GC%, fungi). | Lysis efficiency bias and bioinformatic classification accuracy across taxa. |
Q3: We observe batch-to-batch variation in mock community results even with the same protocol. What could be the issue?
A: This often points to inconsistencies in pre-extraction handling or reagent degradation.
- Cause 1: Inconsistent storage and handling. Repeated freeze-thaw cycles of the mock community stock can lyse cells differentially.
- Solution: Create single-use aliquots of the mock community. Store at -80°C. Thaw on ice immediately before use.
- Cause 2: PCR inhibitor carryover or degraded PCR reagents.
- Solution: Include a positive control (pure microbial DNA from the mock) and a negative control (water) in your library preparation batch. If the positive control deviates, the issue is downstream of extraction (PCR, sequencing). Monitor Ct values in qPCR steps.
Q4: How can I use mock community data to computationally correct for observed bias in my real samples?
A: Mock communities enable bias modeling but not direct 1:1 correction. The standard approach is to use the mock to inform quality filtering and to apply in silico adjustments cautiously.
- Step 1: Calculate "Recovery Factors" (RF) for each taxon i in your mock community: RFi = (Observed Abundancei / Expected Abundance_i).
- Step 2: Apply a threshold (e.g., RF between 0.1 and 10) to identify taxa your pipeline can quantify reliably. Taxa outside this range in your mock should be flagged as "semi-quantitative" in real samples.
- Step 3: For rigid correction, some tools use the RF to adjust counts, but this assumes bias is consistent across sample matrices, which it often is not.
The Scientist's Toolkit: Key Research Reagent Solutions
| Item | Function & Importance |
|---|---|
| ZymoBIOMICS Microbial Community Standards | Defined, whole-cell mock communities with Gram-positive and Gram-negative bacteria and yeast. Served as a gold standard for benchmarking. |
| BEI Resources Mock Viruses & Prokaryomes | NIST-traceable mock communities for complex microbiome studies, including anaerobes. |
| Pathogen Spike-in Controls (e.g., SIRVs for metatranscriptomics) | Defined RNA transcripts added to lysates to evaluate mRNA enrichment and transcriptomic bias. |
| Inhibitor-Removal Matrices (e.g., PVPP, BSA) | Added to lysis buffer to bind humic acids (soil) or bile salts (stool) that co-purify and inhibit downstream PCR. |
| Internal Spike-in DNA (e.g., Synthetic Pseudomonas sythetica) | A non-biological DNA sequence added post-lysis but pre-extraction to quantify absolute abundance and loss during purification. |
| PCR Duplicate Removal Enzymes (e.g., Uracil-DNA glycosylase) | Critical for reducing amplification bias in low-biomass mock community analysis to prevent overestimation of diversity. |
Experimental Protocols
Protocol 1: Comprehensive Method Benchmarking Using a Staggered Mock Community Objective: To evaluate the complete workflow from lysis to bioinformatics.
- Sample Prep: Use a commercially available staggered abundance mock community (e.g., ZymoBIOMICS D6300).
- Lysis Methods in Parallel: Process aliquots with: a) Kit protocol (enzymatic); b) Enhanced protocol (enzymatic + 2x bead beating); c) High-temperature chemical lysis.
- Extraction: Use the same spin-column purification for all lysates.
- Library Prep: Use a dual-indexing 16S rRNA gene (V4) or shotgun metagenomics kit. Include a no-template control.
- Sequencing: Run on a mid-output flow cell (2x150bp) to achieve >100,000 reads per sample.
- Analysis: Process through standard pipeline (DADA2/QIIME2 for 16S; KneadData/MetaPhlAn for shotgun). Compare relative abundances to the provided gold standard ratio sheet.
Protocol 2: Evaluating Matrix Inhibition with Spiked Mock Communities Objective: To assess how a sample matrix affects microbial recovery.
- Matrix Collection: Obtain the target matrix (e.g., stool, soil) confirmed to be negative for your mock community taxa.
- Spiking: Create two sets: A) Pure mock community (reference). B) Mock community spiked into the matrix (e.g., 10^6 cells into 200mg of matrix).
- Extraction: Process both sets with your standard and an "inhibitor-resistant" kit (often with additional wash steps).
- qPCR: Quantify total bacterial 16S rRNA gene copies in all extracts. Calculate percent recovery from the spiked matrix vs. the pure mock.
- Sequencing: Analyze compositional skew.
Visualizations
Title: Mock Community Workflow for Bias Identification
Title: Thesis-Driven Method Evaluation Logic
Troubleshooting Guides & FAQs
Q1: My DNA yield is consistently low from complex environmental samples (e.g., soil, stool). What are the primary causes and solutions? A: Low yield is often due to inefficient cell lysis or analyte loss during purification.
- Cause 1: Inefficient lysis of robust cell walls (e.g., Gram-positive bacteria, spores).
- Solution: Implement a multi-modal mechanical lysis step. Combine bead-beating (0.1mm silica/zirconia beads) with a pre-incubation step using a lytic enzyme cocktail (e.g., lysozyme, mutanolysin, lysostaphin) tailored to your expected community. Increase bead-beating duration in 30-second increments, with cooling intervals to prevent thermal degradation.
- Cause 2: DNA adsorption to co-extracted organic matter (humic acids, polyphenols).
- Solution: Modify your purification buffer. Increase the concentration of a competitive binder like polyvinylpyrrolidone (PVP) to 1-2% w/v. Use silica-membrane columns specifically designed for inhibitor removal, or switch to a CTAB-based precipitation protocol for heavily contaminated samples.
- Protocol - Enhanced Lysis for Low Yield:
- Suspend 0.25g sample in 500µL of enzymatic lysis buffer (20 mM Tris-Cl pH 8.0, 2 mM EDTA, 1.2% Triton X-100, 20 mg/mL lysozyme). Incubate at 37°C for 45 min.
- Add 0.3g of 0.1mm beads and 500µL of a chaotropic binding buffer (e.g., from a commercial kit).
- Bead-beat at 6.5 m/s for 90 seconds. Cool on ice for 2 minutes. Repeat once.
- Proceed with purification, incorporating a wash step with 5 mM ammonium acetate in 80% ethanol to remove residual humics.
Q2: I achieve high DNA yield and purity (A260/A280), but my 16S rRNA gene sequencing results show poor taxonomic fidelity, skewing toward Gram-negative bacteria. How do I correct this? A: This is a classic bias issue from overly aggressive lysis. High yield often comes from complete lysis of easy-to-lyse cells, destroying fragile ones.
- Cause: Differential lysis efficiency disproportionately fragments cells from Gram-positive taxa and delicate lineages.
- Solutions:
- Standardize Lysis Intensity: Calibrate mechanical lysis energy. For bead-beating, optimize to a single, short burst (e.g., 30 seconds at 5 m/s). Validate by microscopy or qPCR for balanced amplification of Gram-positive vs. Gram-negative marker genes.
- Employ a Gentle Protocol: For liquid samples (e.g., saliva, plankton), use a gentle enzymatic/chemical lysis (e.g., SDS-proteinase K at 50°C for 1h) without bead-beating. Compare results to a standardized harsh lysis to understand your bias landscape.
- Use an Internal Spike-in Control: Add a known quantity of cells from an exotic, non-native organism (e.g., Aliivibrio fischeri to soil) prior to lysis. Calculate its recovery rate via qPCR with a unique primer set to quantify lysis bias quantitatively.
Q3: My DNA has low integrity (degraded) and fails long-amplicon PCR. How can I prevent shearing and degradation? A: Degradation occurs via physical shearing or nuclease activity.
- Cause 1: Overly vigorous physical handling during and after lysis.
- Solution: Avoid vortexing after lysis. Perform only gentle inversions. Use wide-bore pipette tips for all post-lysate transfers. Do not freeze-thaw DNA extracts; store at 4°C (short-term) or -80°C in TE buffer.
- Cause 2: Endogenous or contaminating nuclease activity.
- Solution: Ensure lysis buffers contain sufficient chelating agents (EDTA ≥ 5 mM) to inhibit metal-dependent nucleases. Include a step to inactivate nucleases, such as a 65°C incubation for 10 minutes after lysis, or use a denaturing guanidinium thiocyanate buffer from the start.
Q4: How do I systematically choose an extraction kit for my specific sample type to maximize taxonomic fidelity? A: Perform a comparative audit using a mock microbial community.
- Protocol - Kit Evaluation for Fidelity:
- Acquire a Mock Community: Use a commercially available, defined genomic mock community (e.g., from ZymoBIOMICS, ATCC) with known, even abundances.
- Parallel Extraction: Subject identical aliquots of the mock community to 3-4 different extraction kits/methods (e.g., one harsh mechanical, one gentle enzymatic, one phenol-chloroform based).
- Sequencing & Analysis: Perform 16S rRNA gene amplicon sequencing (or shotgun metagenomics) on all extracts in the same sequencing run.
- Metric Calculation: Compare the observed relative abundances to the known template abundances. The kit with the lowest aggregate deviation (e.g., lowest Bray-Curtis dissimilarity) offers the highest taxonomic fidelity for that community type.
Table 1: Comparison of Common DNA Extraction Methods on a ZymoBIOMICS Microbial Community Standard (Even Composition)
| Extraction Method | Avg. Yield (ng) | A260/A280 | A260/A230 | Observed G+/G- Ratio* | Deviation from Expected (BC Dissimilarity) |
|---|---|---|---|---|---|
| Harsh Bead-Beating (Kit A) | 45.2 ± 5.1 | 1.92 ± 0.03 | 2.15 ± 0.10 | 0.6:1 | 0.41 ± 0.05 |
| Gentle Enzymatic (Kit B) | 18.7 ± 2.3 | 1.88 ± 0.05 | 1.95 ± 0.15 | 1.8:1 | 0.28 ± 0.03 |
| Phenol-Chloroform (Manual) | 62.1 ± 7.8 | 1.80 ± 0.10 | 1.70 ± 0.20 | 1.1:1 | 0.35 ± 0.04 |
| Modified Protocol (Kit A + Gentle Lysis) | 32.5 ± 3.4 | 1.90 ± 0.04 | 2.05 ± 0.12 | 1.2:1 | 0.19 ± 0.02 |
Based on 16S rRNA gene amplicon sequencing. Ideal expected ratio for this standard is ~1:1. *Bray-Curtis Dissimilarity between observed and expected profile. Lower is better (0 = perfect fidelity).
Table 2: Impact of Bead-Beating Duration on DNA Metrics from Gram-Positive Rich Soil
| Bead-Beating Time (sec) | Total Yield (ng) | % Yield >10kb* | Firmicutes:Actinobacteria Ratio |
|---|---|---|---|
| 30 | 1050 ± 120 | 85% | 1.5:1 |
| 60 | 2200 ± 250 | 75% | 1.1:1 |
| 120 | 3100 ± 400 | 45% | 0.7:1 |
| 180 | 3200 ± 350 | 20% | 0.5:1 |
*Assessed by Fragment Analyzer; indicates integrity.
Diagrams
Diagram Title: DNA Extraction Workflow & Bias Introduction Point
Diagram Title: Lysis Method Impact on Community Representation
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Bias-Aware DNA Extraction
| Item | Function in Addressing Bias | Example Product/Chemical |
|---|---|---|
| Mock Microbial Community | Gold-standard control for quantifying taxonomic fidelity in extraction and sequencing. | ZymoBIOMICS Microbial Community Standard, ATCC MSA-1003 |
| Inhibitor Removal Additives | Binds to humic acids/polyphenols, improving purity and downstream PCR efficiency from complex matrices. | Polyvinylpyrrolidone (PVP), Bovine Serum Albumin (BSA), PTB |
| Lytic Enzyme Cocktail | Targeted, gentle digestion of specific cell wall types (e.g., Gram-positive) to balance lysis efficiency. | Lysozyme, Mutanolysin, Lysostaphin, Proteinase K |
| Uniform Beads (0.1mm) | Provides consistent mechanical shearing force for cell disruption; size critical for efficiency. | Zirconia/Silica beads (0.1mm for bacteria, 0.5mm for fungi) |
| Internal DNA Spike-in | Distinguishes between lysis bias and purification bias by quantifying recovery of an added control. | gBlock gene fragment, genomic DNA from Aliivibrio fischeri |
| Wide-Bore Pipette Tips | Prevents shearing of high-molecular-weight DNA, preserving integrity for long-read sequencing. | Any certified wide-bore/ low-retention tip |
| Guanidinium Thiocyanate Buffer | Powerful chaotropic agent that denatures proteins/nucleases immediately upon lysis, stabilizing nucleic acids. | Component of many commercial kits (e.g., Qiagen RLT) |
Technical Support Center: Troubleshooting Bias in Microbial DNA Extraction
FAQs & Troubleshooting Guides
Q1: My DNA yield is high, but my 16S rRNA gene sequencing shows low diversity and skews heavily toward a single phylum (e.g., Firmicutes). What is the likely cause and how can I fix it? A: This is a classic sign of lysis bias. Mechanical disruption methods (e.g., bead beating) are often insufficient for robust Gram-positive bacterial cell wall disruption, while enzymatic lysis may preferentially lyse Gram-negative bacteria.
- Solution: Implement a standardized, rigorous mechanical lysis protocol. Increase bead-beating intensity and/or duration. Validate lysis efficiency using microscopy (live/dead staining) or spike-in controls (e.g., known quantities of Bacillus subtilis spores). Consider a multi-enzyme cocktail (lysozyme, mutanolysin, lysostaphin) in combination with mechanical disruption.
Q2: I observe inconsistent community profiles between technical replicates when extracting from the same homogenized sample. What step is most prone to this variability? A: Inconsistent profiles often stem from incomplete sample homogenization prior to subsampling or from the nucleic acid binding step during purification.
- Solution:
- Homogenization: Use a validated, vigorous homogenization method (e.g., vortexing with garnet beads, commercial homogenizers) for at least 2 minutes before aliquoting.
- Binding: Ensure consistent pellet disruption after lysis. For silica-column protocols, verify the pH and salt concentration of the binding buffer is correct. For magnetic bead protocols, ensure consistent mixing during the binding incubation and that beads are not being lost during wash steps.
Q3: How can I determine if my DNA extraction kit is selectively losing extracellular DNA (eDNA) or DNA from lysed cells, which might represent an important fraction of the community? A: This requires a controlled experiment.
- Protocol:
- Generate a mock community with both intact cells (e.g., E. coli, S. aureus) and purified genomic DNA from a distinct organism (e.g., P. aeruginosa).
- Split the mock community. Treat one aliquot with an enzyme that degrades free DNA (e.g., DNase I) prior to extraction; leave the other untreated.
- Extract both aliquots using your protocol and quantify the target DNA from the "free-DNA" organism via qPCR.
- A significant drop in its signal in the DNase-treated sample vs. the untreated sample indicates your protocol is effectively capturing eDNA. A minimal drop suggests your protocol is losing eDNA.
Q4: My negative extraction controls are consistently showing low-level contamination. Which reagents are the most common culprits? A: Enzyme mixes (e.g., lysozyme, proteinase K) and PCR-grade water are frequent sources of low-biomass contamination.
- Troubleshooting Steps:
- Test all reagents individually by adding them to a mock extraction (no sample) and performing a broad-range 16S rRNA PCR followed by sequencing.
- Aliquot enzymes, freeze, and use single aliquots to reduce cross-contamination.
- Include multiple negative controls (reagent-only) in every extraction batch to identify and bioinformatically filter contaminant ASVs/OTUs using tools like
decontam(R package).
Table 1: Consensus Findings from DNA Extraction Benchmarking Studies
| Finding Category | Key Consensus | Supporting Evidence (Generalized) |
|---|---|---|
| Lysis Efficiency | Mechanical disruption (bead beating) is superior to enzymatic or chemical-only methods for overall diversity representation. | Studies show 10-40% higher Shannon diversity and increased detection of Gram-positive taxa (Firmicutes, Actinobacteria) with bead beating. |
| Inhibition Removal | Inclusion of a dedicated inhibitor removal step (e.g., PTB, PVPP) is critical for samples from soil, feces, or blood. | Improves PCR success rate from ~70% to >95% in inhibitor-rich samples and increases library preparation efficiency. |
| Protocol Standardization | Strict adherence to a single, detailed protocol reduces inter-lab variability more than the choice of any specific commercial kit. | Meta-analyses show within-study reproducibility (Beta-diversity distance) is significantly higher than between-study reproducibility for the same sample type. |
| Bias Awareness | No single extraction method is completely unbiased; the "best" method is sample-type and research-question specific. | All benchmark studies conclude that method choice introduces measurable and significant variation in downstream compositional data. |
Table 2: Contradictions and Variability in Published Benchmarks
| Contradiction Point | Divergent Finding A | Divergent Finding B | Potential Reason for Divergence |
|---|---|---|---|
| Kit Performance | Kit "X" consistently yields the highest diversity from human stool. | Kit "Y" outperforms Kit "X" for soil and sediment samples. | Sample matrix profoundly affects reagent efficiency (inhibitor load, humic acids, mucins). |
| Bead Beating Intensity | High intensity/long duration is essential for complete lysis. | Excessive bead beating shears DNA and causes bias against fragile cells/protozoa. | Different sample types have different mechanical resilience optima. Lack of standardized "intensity" metrics. |
| Carrier RNA Efficacy | Carrier RNA (e.g., poly-A) is vital for high yields from low-biomass samples. | Carrier RNA can introduce batch-specific contamination and should be avoided. | Trade-off between sensitivity and contamination risk. Critical in ultra-low-biomass studies (e.g., tissue, air). |
Experimental Protocol: Standardized Bead-Beating for Microbial Community Analysis
Objective: To minimize lysis bias in complex, tough-to-lyse samples (e.g., soil, biofilm, stool). Reagents: Lysis Buffer (e.g., Tris-EDTA-SDS), Proteinase K (20 mg/mL), Lysozyme (100 mg/mL), 0.1 mm and 0.5 mm zirconia/silica beads, Binding Buffer, Wash Buffers, Elution Buffer. Equipment: Vortex adapter for tubes or a homogenizer (e.g., MagNA Lyser, Bead Ruptor), Microcentrifuge, Heated shaker/block.
Methodology:
- Weigh 100-250 mg of sample into a 2 mL screw-cap tube.
- Add 750 µL of pre-warmed (55°C) Lysis Buffer, 30 µL of Proteinase K, and 50 µL of Lysozyme.
- Add a mixture of 0.1 mm (100 mg) and 0.5 mm (200 mg) beads.
- Securely cap tubes and place in a vortex adapter. Vortex at maximum speed for 10 minutes.
- Incubate at 55°C for 15 minutes with shaking (900 rpm).
- Centrifuge at 13,000 x g for 1 minute to pellet beads and debris.
- Transfer supernatant (~600 µL) to a new tube.
- Add 1 volume of Binding Buffer, mix, and proceed with your chosen silica-column or magnetic-bead purification protocol.
- Elute DNA in 50-100 µL of pre-warmed (55°C) Elution Buffer or TE Buffer.
The Scientist's Toolkit: Key Reagent Solutions
| Item | Function & Rationale |
|---|---|
| Zirconia/Silica Beads (0.1 & 0.5 mm mix) | Provides efficient mechanical shearing for diverse cell wall types. The mix targets both small and large cells. |
| Inhibitor Removal Matrix (e.g., PTB, PVPP) | Binds to humic acids, polyphenols, and other common PCR inhibitors co-extracted from environmental/clinical samples. |
| Carrier RNA (Poly-A) | Enhances binding of minute quantities of microbial DNA to silica membranes/beads, improving yield from low-biomass samples. Must be nuclease-free. |
| Multi-Enzyme Lysis Cocktail | Combination of lysozyme (Gram+), mutanolysin (Gram+), and lysostaphin (Staphylococcus) ensures broad enzymatic lysis to complement mechanical disruption. |
| Internal DNA Spike-in Control (e.g., Pseudomonas gDNA) | A known quantity of DNA from an organism absent in the native sample, added pre-extraction, to normalize for extraction efficiency and identify inhibition. |
Visualizations
Troubleshooting Bias in Microbial Community Analysis
Optimal DNA Extraction Workflow for Bias Reduction
Statistical Tools for Bias Assessment and Correction in Post-sequencing Data Analysis
Technical Support Center: Troubleshooting Guides & FAQs
Q1: My negative control shows high read counts, suggesting contamination. What statistical tools can help identify if this is significant bias?
A: Use a combination of prevalence and abundance-based filters.
- Tool Recommendation: Decontam (R package) using its prevalence method.
- Protocol:
- Create a feature table (ASV/OTU counts) and a corresponding sample metadata vector marking true samples (FALSE) and negative controls (TRUE).
- Run the
isContaminant()function withmethod="prevalence"and a reasonable threshold (e.g.,threshold=0.5). This identifies taxa that are significantly more prevalent in negative controls. - Statistically, it performs a Fisher's exact test for each taxon, comparing its presence/absence frequency between true samples and controls.
- Remove flagged contaminants from your dataset.
Q2: After using a compositionality-aware tool like ANCOM-BC, my differential abundance results are empty. What went wrong?
A: This often stems from overly conservative correction for multiple testing or insufficient statistical power.
- Troubleshooting Steps:
- Check the
p_adj_methodargument: Default is often FDR (Benjamini-Hochberg). Try a less stringent method like "BH" or review raw p-values (p_val) for trends. - Review the
lib_cutandstruc_zeroparameters: Increasinglib_cutexcludes low-depth samples, which can improve model stability. Thestruc_zerodetection may have removed taxa, reducing findings. - Power Issue: For low-biomass or highly variable communities, even tools correcting for bias may lack power. Ensure sufficient biological replicates (n>=5 per group is a common minimum).
- Check the
Q3: When using SILVA/Green genes databases for taxonomic assignment, I observe a strong bias against a specific phylum. How can I assess and correct this?
A: This is likely a reference database bias. Assessment requires a mock community experiment.
- Assessment Protocol:
- Experiment: Sequence a commercially available, defined mock community (e.g., ZymoBIOMICS, ATCC MSA-1003) alongside your samples using the identical wet-lab and bioinformatic pipeline.
- Analysis: Use a bias assessment table (see Table 1).
- Correction Tools: Post-hoc, consider using SourceTracker or Decontam (frequency method) if you have mock community data as "controls" to identify taxa with biased recovery.
Q4: My alpha diversity metrics (Chao1, Shannon) vary drastically between different sequencing depths after rarefaction. How do I stabilize results?
A: This indicates sensitivity to sampling depth bias. Avoid single rarefaction.
- Tool Recommendation: Use breakaway for species richness (Chao1 alternative) or Hill numbers calculated from a bootstrapped, smoothed rarefaction curve.
- Protocol for Bootstrapped Hill Numbers:
- Use the
iNEXTR package. - Input your untrimmed feature table and sample metadata.
- Run
iNEXT()withq=c(0,1,2)for Hill numbers corresponding to richness, Shannon exp, and Simpson indices. - The function uses rarefaction and extrapolation based on a unified framework, providing stable estimates with confidence intervals, mitigating the bias of a single rarefaction depth.
- Use the
Q5: How can I statistically differentiate between technical bias from DNA extraction and true biological variation?
A: Implement a replicated extraction design and use PERMANOVA with a nested model.
- Experimental Design: For a subset of biological samples, perform multiple independent DNA extractions (technical replicates).
- Statistical Protocol:
- Generate a beta-diversity distance matrix (e.g., Aitchison for compositionality).
- Use
adonis2in vegan (R) with the formula:distance ~ Biological Condition + Biological Sample ID. Here, 'Biological Sample ID' is nested within 'Condition'. - The variance explained by 'Biological Sample ID' (the extraction effect) versus 'Condition' quantifies the relative impact of technical bias. A significant 'Condition' term after accounting for extraction noise suggests robust biological signal.
Key Research Reagent Solutions
| Item | Function in Bias Assessment/Correction |
|---|---|
| ZymoBIOMICS Microbial Community Standard | Defined mock community of bacteria/fungi. Serves as a positive control to quantify taxonomic abundance bias and batch effects. |
| DNase/RNase-Free Water | Used for negative extraction and PCR controls. Essential for identifying kit/lab-derived contamination. |
| Internal Spike-in DNA (e.g., Synthetic dsDNA, pBI143) | Known quantity of non-biological DNA added pre-extraction. Enables absolute quantification and corrects for yield bias. |
| Inhibitor Removal Technology Beads (e.g., Zymo OneStep PCR Inhibitor Removal) | Reduces co-purified inhibitors that cause biased PCR amplification, improving diversity representation. |
| Magnetic Stand for Bead Cleanup | Standardizes the post-PCR cleanup step, reducing technical variation in final library concentrations. |
Table 1: Mock Community Analysis for Database Bias Assessment
| Taxon (Expected) | Expected Rel. Abundance (%) | Observed Rel. Abundance (%) | Bias Fold-Change (Obs/Exp) |
|---|---|---|---|
| Pseudomonas aeruginosa | 12.5 | 18.7 | 1.50 |
| Escherichia coli | 12.5 | 9.8 | 0.78 |
| Bacillus subtilis | 12.5 | 5.2 | 0.42 |
| Lactobacillus fermentum | 12.5 | 15.1 | 1.21 |
Table 2: Common Statistical Tools for Bias Correction
| Tool/Method | Primary Bias Addressed | Key Principle | Output |
|---|---|---|---|
| ANCOM-BC | Compositionality, Sampling Fraction | Linear model with bias correction term for log-ratios. | Corrected log-fold changes, p-values. |
| Decontam (prevalence) | Contamination | Fisher's exact test on prevalence in samples vs. controls. | Logical vector of contaminant IDs. |
| DESeq2 (with caution) | Library Size, Variance | Negative binomial GLM with variance stabilization. | Normalized counts, differential abundance. |
| GUniFrac | Phylogenetic, Sampling Depth | Generalized UniFrac distance incorporating abundance. | Distance matrix less sensitive to depth. |
| Rarefaction with Bootstrapping | Unequal Sampling Depth | Repeated subsampling to generate stable estimates. | Smoothed diversity curves with CIs. |
Experimental Protocols
Protocol 1: Assessing Extraction Bias Using a Mock Community
Objective: Quantify taxon-specific biases introduced by the DNA extraction protocol.
- Materials: ZymoBIOMICS Microbial Community Standard, chosen DNA extraction kit, internal spike-in DNA (optional).
- Procedure: a. Replicate Extractions: Perform the extraction protocol on n≥5 aliquots of the mock community standard. b. Sequencing: Process all extracts through the same 16S/ITS rRNA gene amplicon or shotgun sequencing pipeline alongside experimental samples. c. Bioinformatic Processing: Process raw reads through standard pipeline (DADA2, QIIME2) to obtain a feature table. d. Statistical Analysis: Generate Table 1. Calculate correlation (e.g., Spearman's ρ) between expected and observed abundances. Perform a Wilcoxon test on Bias Fold-Change values to see if they significantly deviate from 1.
Protocol 2: Implementing a Nested Design for Bias Partitioning
Objective: Statistically partition variance between biological condition and technical extraction noise.
- Experimental Design: Select 3 biological samples per condition. For each biological sample, conduct 3 independent DNA extractions (technical replicates).
- Wet-Lab & Sequencing: Process all extracts (biological x technical) through library prep and sequencing in a randomized batch.
- Bioinformatics: Generate an Aitchison distance matrix from centered log-ratio (CLR) transformed counts.
- Statistical Testing: Use Permutational Multivariate Analysis of Variance (PERMANOVA) with a nested model in R:
Visualizations
Diagram Title: Statistical Bias Correction Workflow in Sequencing Analysis
Diagram Title: Nested Experimental Design for Bias Partitioning
Technical Support Center: Troubleshooting Bias in Microbial DNA Extraction
FAQs & Troubleshooting Guides
Q1: Our 16S rRNA sequencing results show very low biomass for Gram-positive bacteria compared to Gram-negative controls. What could be the cause? A: This is a classic sign of extraction bias. Gram-positive bacteria have thick, rigid peptidoglycan cell walls that are resistant to mechanical and chemical lysis. If your protocol is optimized for Gram-negatives, it may under-lyse Gram-positives.
- Troubleshooting Steps:
- Add a enzymatic pre-treatment: Incorporate a 30-minute incubation at 37°C with lysozyme (20 mg/mL) and/or mutanolysin prior to the main lysis step.
- Increase mechanical disruption: If using bead beating, ensure you are using a homogenizer (e.g., FastPrep, Bead Ruptor) with appropriate speed and time (e.g., 6.0 m/s for 45-60 seconds). Validate with a mix of known Gram-positive and Gram-negative cells.
- Review chemical lysis: Ensure your lysis buffer contains sufficient concentrations of chaotropic agents (e.g., guanidine thiocyanate) and detergents (e.g., SDS).
Q2: We observe high variation in community profiles between technical replicates from the same soil sample. How can we improve consistency? A: High technical variation often stems from incomplete or inconsistent homogenization of the starting material, especially for complex, heterogeneous samples like soil or stool.
- Troubleshooting Steps:
- Standardize sample homogenization: For soil/stool, use a dedicated bench-top homogenizer (e.g., PowerLyzer) for a fixed duration. For biofilms, consider a vortex adapter for tubes.
- Increase sample input mass: If possible, increase the starting material mass to average out heterogeneity, provided your kit's binding capacity is not exceeded.
- Implement a positive control: Spike in a known quantity of an exogenous control (e.g., Pseudomonas aeruginosa or a synthetic DNA spike) to monitor and normalize for extraction efficiency variance.
Q3: Our metagenomic data seems to be contaminated with human host DNA, overwhelming the microbial signal. How can we mitigate this? A: This is common in host-associated samples (e.g., saliva, tissue). Selective lysis or post-extraction depletion can help.
- Troubleshooting Steps:
- Selective lysis: Use a gentle lysis step (e.g., weak detergents like Triton X-100) to open mammalian cells, digest DNA with a DNase, then proceed with harsh microbial lysis.
- Commercial depletion kits: Employ host DNA depletion kits (e.g., NEBNext Microbiome DNA Enrichment Kit, QIAamp DNA Microbiome Kit) that use methylation-dependent restriction enzymes or probe capture.
- Protocol Adjustment: For tissue, include a physical separation step (e.g., centrifugation) to pellet microbial cells away from host cell debris before lysis.
Q4: How do we know if our extraction protocol is introducing significant bias, and what benchmarks should we use? A: You must validate your protocol against a mock microbial community with a known, even composition. Performance metrics should be quantitatively tracked.
- Validation Protocol:
- Obtain a Mock Community: Use a commercially available, sequenced mock community (e.g., from ZymoBIOMICS, ATCC, BEI Resources) containing both Gram-positive and Gram-negative bacteria, as well as fungi if relevant.
- Extract in Triplicate: Perform your standard extraction protocol on the mock community.
- Sequence and Analyze: Perform 16S rRNA gene sequencing (using a standardized primer set, e.g., 515F/806R for V4) or shotgun metagenomics. Compare the observed relative abundances to the known truth via metrics like Relative Error and Bray-Curtis Dissimilarity.
Quantitative Data Summary: Comparison of Common Extraction Kits Against a Mock Community
Table 1: Performance metrics for three common DNA extraction methods tested on the ZymoBIOMICS Microbial Community Standard (D6300). Data is illustrative of typical peer-reviewed findings.
| Extraction Method | Type | Mean Relative Error (%) | Bray-Curtis Dissimilarity vs. Expected | DNA Yield (ng) | Notable Bias |
|---|---|---|---|---|---|
| Kit A (Enzymatic + Bead Beat) | Bead-beating intensive | 8.5 | 0.12 | 45.2 | Minimal; best for tough Gram-positives. |
| Kit B (Spin Column, gentle lysis) | Chemical lysis primary | 32.1 | 0.41 | 28.7 | Severe under-representation of Gram-positives. |
| Kit C (Phenol-Chloroform + Beads) | Manual/organic | 15.3 | 0.19 | 62.5 | Slight bias towards high-GC organisms. |
Experimental Protocol: Comprehensive Bias Assessment
Title: Protocol for Assessing DNA Extraction Bias Using a Mock Microbial Community.
Objective: To quantitatively evaluate the bias introduced by a DNA extraction protocol on microbial community representation.
Materials: See "The Scientist's Toolkit" below.
Procedure:
- Sample Preparation: Resuspend the lyophilized mock microbial community standard according to the manufacturer's instructions. Aliquot equal volumes (e.g., 200 µL) into 6-8 sterile 2 mL screw-cap tubes.
- Extraction: Perform your test DNA extraction protocol on triplicate aliquots. Include one replicate of a validated, high-performance kit (e.g., Kit A from Table 1) as a positive control.
- Negative Control: Include a tube containing only sterile PBS or nuclease-free water processed through the entire extraction protocol.
- DNA Quantification & QC: Quantify DNA yield using a fluorescent assay (e.g., Qubit dsDNA HS Assay). Assess quality via absorbance ratios (A260/A280 ~1.8, A260/A230 >2.0) and fragment analyzer.
- Library Preparation & Sequencing: Prepare 16S rRNA gene amplicon libraries for the V4 region using dual-indexed primers (e.g., 515F/806R). Pool libraries equimolarly and sequence on an Illumina MiSeq (2x250 bp).
- Bioinformatic Analysis:
- Process sequences using DADA2 or QIIME 2 to generate amplicon sequence variants (ASVs).
- Classify ASVs against a reference database (e.g., SILVA).
- Bias Calculation:
- Calculate Relative Error for each taxon:
(Observed Abundance - Expected Abundance) / Expected Abundance * 100%. - Calculate Bray-Curtis Dissimilarity between the observed composition vector and the expected composition vector.
- Calculate Relative Error for each taxon:
Mandatory Visualizations
Title: Microbial DNA Extraction Workflow & Bias Introduction Points
Title: Consequences of Opaque Method Reporting in Publications
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential materials for transparent and robust microbial DNA extraction research.
| Item | Function/Justification | Example Product/Catalog |
|---|---|---|
| Characterized Mock Community | Gold-standard for quantifying extraction bias and benchmarking. | ZymoBIOMICS Microbial Community Standard (D6300) |
| Inhibitor-Removal Technology | Critical for samples like soil or stool; improves PCR success. | OneStep PCR Inhibitor Removal Kit (Zymo Research) |
| High-Efficiency Beads | Consistent mechanical lysis. A mix of sizes (e.g., 0.1 & 0.5mm) improves efficiency. | Garnet beads, 0.1mm (OMNI International) |
| Lysozyme & Mutanolysin | Enzymatic pre-treatment for effective Gram-positive lysis. | Lysozyme (Sigma L4919), Mutanolysin (Sigma M9901) |
| Fluorometric DNA Quant Assay | Accurate quantification of double-stranded DNA, unaffected by contaminants. | Qubit dsDNA HS Assay Kit (Thermo Fisher) |
| Exogenous Internal Control | Synthetic DNA spike-in to monitor extraction efficiency and normalize yields. | Spike-in Control (e.g., from ATCC MSA-1002) |
| Standardized Lysis Buffer | Ensures consistent chemical lysis. MO BIO (now Qiagen) PowerSoil Bead Solution is a common base. | PowerBead Solution (Qiagen) |
Conclusion
DNA extraction bias is not a mere technical nuisance but a fundamental challenge that underpins the interpretability and reproducibility of all downstream microbiome analyses. A robust strategy requires understanding the sources of bias (Intent 1), strategically selecting a fit-for-purpose method (Intent 2), meticulously optimizing it for the specific sample matrix (Intent 3), and rigorously validating its performance against known standards (Intent 4). For biomedical and clinical research, where microbiome signatures are being explored as diagnostics, therapeutics, and biomarkers, acknowledging and mitigating this bias is non-negotiable. Future directions must move towards community-accepted standardized protocols for key sample types, the development of novel extraction chemistries that minimize differential lysis, and the broader adoption of internal spike-in controls to enable quantitative and cross-study comparisons. Only by confronting the bias at the very first step can we build a truly accurate and reliable picture of the microbiome's role in health and disease.