Granger Causality in Ecological Interaction Networks: A Comprehensive Guide for Biomedical Researchers
This article provides a detailed exploration of Granger causality (GC) analysis for inferring directed interactions in complex ecological networks, with a focus on biomedical applications.
Granger Causality in Ecological Interaction Networks: A Comprehensive Guide for Biomedical Researchers
Abstract
This article provides a detailed exploration of Granger causality (GC) analysis for inferring directed interactions in complex ecological networks, with a focus on biomedical applications. We first establish the foundational concepts of GC and its adaptation from econometrics to microbial and host-ecosystem studies. Next, we delve into methodological implementations—from classic vector autoregression (VAR) to modern nonlinear and conditional approaches—and their application in microbiome, multi-omics, and disease-state network reconstruction. We then address common pitfalls, such as stationarity requirements, confounder management, and computational optimization for high-dimensional data. Finally, we compare GC to alternative network inference methods (e.g., correlation, Bayesian networks, transfer entropy), evaluating their strengths and validation frameworks. This guide is tailored for researchers and drug development professionals seeking to uncover causal dynamics in ecological systems relevant to human health.
What is Granger Causality? From Economic Time Series to Ecological Network Inference
Application Notes: Granger Causality in Ecological Interaction Networks
Granger causality (GC) is a statistical hypothesis test for determining whether one time series is useful in forecasting another. Within ecological interaction networks research, it provides a formal framework for inferring predictive precedence—the idea that if a signal X "Granger-causes" Y, then past values of X contain information that helps predict Y above and beyond the information contained in past values of Y alone. This is pivotal for disentangling directional influences in complex systems like species abundance dynamics, gene regulatory networks in microbial communities, or host-pathogen-vector interactions.
Core Principle: Predictive Precedence, Not True Causality. GC identifies predictive relationships from observational data. It does not establish mechanistic causality but indicates that one variable precedes and provides statistically significant information about another. This is particularly valuable in ecology, where controlled manipulativse experiments are often impossible.
Key Assumptions for Ecological Application:
- Temporal Data: Variables must be measured as time series.
- Stationarity: The joint probability distribution of the variables should be invariant to shifts in time.
- Causal Sufficiency: All relevant variables influencing the system are included in the model.
Recent Advancements (2023-2024): Modern applications address traditional limitations through:
- Convergent Cross Mapping (CCM): Used alongside GC to test for dynamical coupling in nonlinear systems.
- Conditional & Multivariate GC: Isolates direct causality in networks with multiple interacting species or environmental factors.
- Frequency-Domain GC: Uncovers causal relationships at specific timescales (e.g., seasonal vs. annual cycles).
Table 1: Comparison of Granger Causality Methods in Ecological Research
| Method | Key Feature | Best Used For | Key Limitation Addressed |
|---|---|---|---|
| Bivariate GC | Tests relationship between two variables. | Preliminary screening of pairwise interactions. | Confounding by latent variables. |
| Conditional GC | Tests if X causes Y, conditioned on a set of other variables Z. | Isolating direct effects in a known network. | Omitted variable bias; requires measuring key covariates. |
| Multivariate GC (Vector Autoregression) | Models all variables simultaneously in a single system. | Full network inference from multivariate time series. | Requires more data; computationally intensive. |
| Frequency-Domain GC | Decomposes causality into specific frequency bands. | Identifying causal cycles (e.g., diurnal, seasonal). | Interpretation complexity; assumes linearity. |
| Nonlinear GC (Kernel-based) | Uses kernel functions to model nonlinear relationships. | Complex species interactions with thresholds/saturations. | High computational cost; risk of overfitting. |
Experimental Protocols
Protocol 2.1: Bivariate Granger Causality Test for Species Abundance Time Series
Objective: To test if the past abundance of Species A predicts the current abundance of Species B.
Materials & Data:
- Time series data for Species A and B abundances (e.g., from camera traps, transect counts, or metabarcoding) collected at regular intervals (t=1, 2, ..., T).
- Statistical software (R, Python with
statsmodels, or specialized packages likeMVGC).
Procedure:
- Data Preprocessing & Stationarity Check:
- Visually inspect time series for trends/seasonality.
- Apply differencing or transformation (e.g., log(x+1)) if necessary.
- Perform Augmented Dickey-Fuller (ADF) test. Requirement: p-value < 0.05 to reject non-stationarity.
Model Specification - Lag Selection:
- Fit Vector Autoregression (VAR) models with lags from 1 to p_max.
- Use Akaike Information Criterion (AIC) to select optimal lag order (p).
Model Estimation:
- Estimate two autoregressive models:
- Restricted Model (R): Y(t) = Σ{i=1}^p αi Y(t-i) + εR(t)
- Unrestricted Model (U): Y(t) = Σ{i=1}^p αi Y(t-i) + Σ{i=1}^p βi X(t-i) + εU(t)
- Estimate two autoregressive models:
Hypothesis Testing (F-test):
- Calculate Residual Sum of Squares (RSS) for both models.
- Compute F-statistic: F = [(RSSR - RSSU) / p] / [RSS_U / (T - 2p - 1)]
- Null Hypothesis (H0): β1 = β2 = ... = β_p = 0 (X does not Granger-cause Y).
- If F-statistic > critical value (or p-value < 0.05), reject H0. Conclude X Granger-causes Y.
Validation:
- Check model residuals for autocorrelation (Ljung-Box test).
- Perform sensitivity analysis on lag selection.
Protocol 2.2: Conditional Granger Causality for Tri-Trophic Interaction
Objective: To determine if predator abundance (C) directly affects prey (B), conditioned on the resource (A) of the prey.
Procedure:
- Data Preparation: Ensure stationary, aligned time series for Resource (A), Prey (B), and Predator (C).
Full and Restricted VAR Models:
- Full Model (F): Includes past values of A, B, and C to predict B(t).
- Restricted Model (R): Includes only past values of A and B to predict B(t).
Conditional F-test:
- Compare the variance explained by the full model versus the restricted model.
- A significant result indicates that C provides predictive information about B beyond what is contained in A and B's own past—suggesting a direct or indirect causal link from predator to prey, conditioned on resource availability.
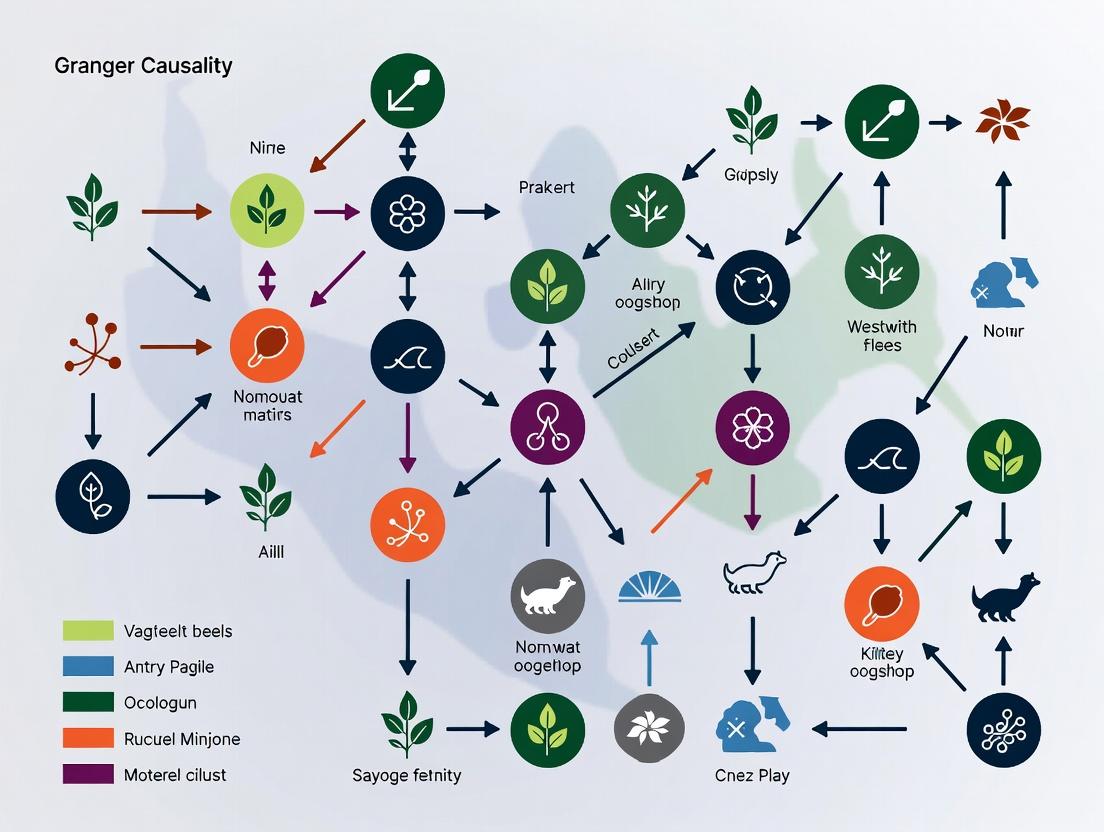
Visualizations
Diagram Title: Granger Causality Testing Protocol Workflow
Diagram Title: GC-Inferred Trophic Cascade with Feedback
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Tools for Granger Causality Analysis in Ecological Networks
| Item / Solution | Function in Research | Example / Note |
|---|---|---|
| High-Frequency Time Series Data | Primary input for GC analysis. Requires consistent temporal resolution. | Remote sensing NDVI, automated acoustic monitors, eDNA sampling time series, long-term ecological survey data. |
| Statistical Software Packages | Implement VAR models, lag selection, and GC tests. | R: vars, lmtest, MVGC packages. Python: statsmodels.tsa.vector_ar, GrangerCausality. |
| Stationarity Testing Suite | Validate a core assumption of GC. | Augmented Dickey-Fuller test (ADF), Kwiatkowski-Phillips-Schmidt-Shin (KPSS) test. |
| Lag Order Selection Criteria | Determine the optimal historical window (p) for the model. | Akaike Information Criterion (AIC), Bayesian Information Criterion (BIC). |
| Conditional GC Algorithm | Isolates direct causality in multi-species networks, controlling for confounding variables. | Implemented in MVGC toolbox. Critical for moving beyond pairwise analysis. |
| Bootstrap Resampling Scripts | Assess the robustness and significance of inferred causal links. | Used to generate empirical null distributions for GC test statistics. |
| Nonlinear GC Extensions | Analyze systems where relationships are not linear. | Kernel-based GC or transfer entropy-based methods (e.g., R TransferEntropy). |
Application Notes: Conceptual Translation & Core Assumptions
The application of Granger causality (GC), a statistical hypothesis test for time-series prediction originating in econometrics, to ecological and biological systems requires careful translation of its core assumptions and adaptation to the unique properties of living systems.
Table 1: Translation of Granger's Framework from Economics to Ecology/Biology
| Aspect | Original Economic Context (Granger, 1969) | Adapted Ecological/Biological Context | Key Considerations & Challenges |
|---|---|---|---|
| Core Definition | Variable X "Granger-causes" Y if past values of X contain information that helps predict Y beyond the information contained in past values of Y alone. | Species/Population X "Granger-causes" Y if past abundance (or state) of X improves prediction of future abundance/state of Y, conditional on Y's own past. | Ecological interactions are often non-linear and non-additive. Feedback loops are the norm. |
| Temporal Resolution | Regular, fixed intervals (e.g., quarterly GDP). | Often irregular or mismatched. May require interpolation or state-space models. | Sampling frequency must capture the relevant dynamical timescales of interaction (e.g., predator-prey cycles). |
| System Stationarity | Assumed (or detrended) for standard tests. | Rarely achieved. Populations trend, oscillate, and undergo regime shifts. | Requires differentiation, detrending, or use of non-stationary GC methods (e.g., time-varying). |
| Causal Sufficiency | Often assumed (no missing confounding variables). | Almost always violated. Unmeasured abiotic factors (climate) or hidden species confound inference. | Must be explicitly acknowledged. Partial GC and conditional GC are essential tools. |
| Interaction Nature | Linear, directional influence. | Non-linear, bidirectional (feedbacks), indirect (through intermediaries), and higher-order. | Linear Vector Autoregression (VAR) models may fail. Kernel or non-parametric GC extensions are needed. |
| Noise Structure | Typically Gaussian. | Complex, potentially non-Gaussian, with measurement error. | Model residuals must be checked. Ecological data often has Poisson or negative binomial distributions. |
Protocols for Granger Causality Analysis in Ecological Networks
Protocol 2.1: Pre-processing and Stationarity Assessment for Ecological Time-Series
Objective: Prepare multivariate ecological time-series data (e.g., species counts, gene expression levels, metabolite concentrations) for GC analysis.
Materials & Software:
- Time-series data matrix (TxN for T time points and N variables).
- Statistical software (R with
vars,lmtest,pscl, orMARSSpackages; Python withstatsmodels,NiTime). - (Optional) State-space modeling package (
MARSSin R).
Procedure:
- Data Alignment: Ensure all time series are on a common, regular time interval. For irregular data, use linear interpolation or Bayesian state-space models to estimate states at regular intervals.
- Missing Data Imputation: Use Kalman filtering (for continuous data) or appropriate imputation methods (e.g., last observation carried forward, EM algorithm) for minor gaps. Extensive missing data requires specialized state-space approaches.
- Trend & Seasonality Removal:
- Visually inspect series for trends and periodic cycles.
- Apply first-differencing (Δyt = yt - y_{t-1}) to remove linear trends.
- For seasonal cycles, use seasonal differencing or include sinusoidal terms as covariates in the subsequent VAR model.
- Stationarity Testing: Perform Augmented Dickey-Fuller (ADF) or Kwiatkowski-Phillips-Schmidt-Shin (KPSS) tests on each processed series. Non-stationary series require further differencing or modeling within a cointegration framework.
- Modeling Count Data: For species abundance counts, consider a Poisson or Negative Binomial VAR model (via
tscountorglmpackages) or transform data (e.g., log(x+1)) with caution regarding interpretation.
Protocol 2.2: Conditional Granger Causality via Vector Autoregression (VAR)
Objective: Test for pairwise and conditional GC within an N-variable system to infer direct interactions.
Procedure:
- Lag Selection: For the full N-variable system, determine the optimal lag length (p) using Akaike Information Criterion (AIC) or Bayesian Information Criterion (BIC) on a series of VAR(p) models.
- Full Model Estimation: Fit a VAR(p) model including all N variables: Y(t) = A1Y(t-1) + ... + ApY(t-p) + ε(t), where Y(t) is the vector of states at time t, Ai are coefficient matrices, and ε is white noise.
- Restricted Model Estimation: To test if X Granger-causes Y, fit a restricted VAR model where the past lags of X are omitted from the equation for Y.
- Hypothesis Testing: Perform a likelihood-ratio test or F-test comparing the full and restricted models for Y's equation. A significant test statistic (p < 0.05, with False Discovery Rate correction for multiple tests) indicates X Granger-causes Y conditional on all other variables in the system.
- Network Inference: Repeat steps 3-4 for all pairwise combinations to construct an N x N adjacency matrix of significant conditional GC relationships. This matrix represents the inferred direct causal network.
Protocol 2.3: Non-Linear Granger Causality using Transfer Entropy
Objective: Detect non-linear causal interactions common in ecological systems.
Procedure:
- Embedding: Reconstruct the state space for each variable using time-delay embedding (Takens' theorem). Determine optimal embedding dimension (m) and delay (τ) using false nearest neighbors and mutual information methods, respectively.
- Transfer Entropy Calculation: Compute the Transfer Entropy (TE) from time series X to Y, which is a model-free, information-theoretic measure equivalent to non-linear GC: TE{X→Y} = Σ p(y{t+1}, yt^{(m)}, xt^{(l)}) log [ p(y{t+1} | yt^{(m)}, xt^{(l)}) / p(y{t+1} | y_t^{(m)}) ] where m and l are embedding dimensions for Y and X.
- Significance Testing: Use a permutation (surrogate) test:
- Randomly shuffle the source series (X) multiple times (e.g., 1000 iterations) to break temporal relationships while preserving distribution.
- Recompute TE for each shuffled surrogate.
- The true TE is significant if it exceeds the (1-α) percentile (e.g., 95th) of the surrogate distribution.
- Conditional TE: To account for common drivers Z, compute conditional TE: TE_{X→Y|Z}. This involves conditioning the probabilities on the embedded states of Z.
Visualization of Methodological Frameworks
Title: GC Analysis Workflow for Ecological Data
Title: Conditional GC Reveals Indirect Effects
The Scientist's Toolkit: Essential Research Reagents & Solutions
Table 2: Key Reagents & Computational Tools for GC Analysis in Biology
| Item/Category | Specific Example/Software | Function in GC Analysis |
|---|---|---|
| Time-Series Data Collection | Long-Term Ecological Research (LTER) datasets; Longitudinal metagenomics/transcriptomics data. | Provides the essential multivariate, temporally ordered observations required for causal inference. |
| Data Pre-processing Suites | R: zoo, imputeTS, forecast. Python: pandas, scipy.signal. |
Handles alignment, interpolation, detrending, and stationarity testing of raw time-series data. |
| Linear GC & VAR Modeling | R: vars, lmtest. Python: statsmodels.tsa.vector_ar. |
Implements core VAR model fitting, lag selection, and linear Granger causality testing. |
| Non-linear GC & Information Theory | R: RTransferEntropy, rneos. Python: JIDT (Java-based). |
Calculates Transfer Entropy and performs non-parametric, non-linear causality testing. |
| State-Space & Bayesian Modeling | R: rstan, MARSS, bsvars. Python: PyMC3, TensorFlow Probability. |
Models latent states, handles missing data, and allows for Bayesian GC inference with uncertainty quantification. |
| Network Visualization & Analysis | R: igraph, qgraph, network. Python: networkx, igraph. Python/Cytoscape. |
Visualizes inferred GC networks and calculates network topology metrics (e.g., connectivity, centrality). |
| High-Performance Computing | Cloud platforms (AWS, GCP), SLURM clusters. | Enables computationally intensive analyses like large-scale permutation testing, high-dimensional GC, and complex simulations. |
Within ecological interaction network research, establishing causal directionality is paramount. Granger causality (GC) provides a statistical framework for inferring directed influence based on temporal precedence and conditional independence. This protocol outlines its application for inferring species interactions, perturbation responses, and network stability, critical for identifying ecological drivers and potential drug targets derived from natural systems.
Foundational Protocols for Granger Causality Analysis
Protocol 2.1: Preprocessing and Stationarity Testing for Ecological Time-Series Data
Objective: Ensure time-series data (e.g., species abundance, metabolite concentration) meets the fundamental assumptions for GC testing. Materials: Ecological time-series dataset (multivariate), statistical software (R/Python). Procedure:
- Data Collection: Assemble longitudinal data for all candidate variables (e.g., n species) at T time points. Sampling frequency must be consistent.
- Detrending & Transformation: Apply logarithmic or square-root transformations to stabilize variance if needed. Remove deterministic trends using polynomial or smooth spline fitting.
- Stationarity Assessment: Perform Augmented Dickey-Fuller (ADF) test on each variable series. Null hypothesis: series is non-stationary.
- Differencing: If non-stationary, apply first-differencing: X'(t) = X(t) - X(t-1). Re-test for stationarity.
- Lag Selection: Use information criteria (AIC/BIC) on Vector Autoregression (VAR) models to determine optimal lag length (p).
Table 1: Stationarity Test Results (Hypothetical Microbial Community)
| Species/Variable | ADF Statistic (Raw) | p-value (Raw) | Stationary? (Raw) | ADF Statistic (Differenced) | p-value (Differenced) | Stationary? (Differenced) |
|---|---|---|---|---|---|---|
| Bacteroides spp. | -1.23 | 0.66 | No | -5.89 | <0.001 | Yes |
| Clostridium spp. | -2.01 | 0.28 | No | -6.45 | <0.001 | Yes |
| Butyrate Concentration | -3.45 | 0.01 | Yes | - | - | - |
| pH | -0.89 | 0.79 | No | -4.12 | <0.001 | Yes |
Protocol 2.2: Bivariate vs. Conditional Granger Causality Testing
Objective: Distinguish direct from indirect causal links by testing conditional independence.
Materials: Stationary multivariate time-series, software with GC toolkits (e.g., statsmodels in Python, lmtest in R).
Procedure for Conditional GC:
- Full Model: Regress the target variable Y(t) on the past of all variables in the set Z (including X). E.g., Y(t) = Σ [α_i * Y(t-i)] + Σ [β_j * X(t-j)] + Σ [γ_k * O(t-k)] + ε(t), where O represents other variables.
- Restricted Model: Regress Y(t) on the past of all variables except X.
- F-test: Compare the residual sum of squares (RSS) of the full and restricted models. The null hypothesis is X does not Granger-cause Y conditional on O.
- Iterate: Repeat for all variable pairs to construct a directed adjacency matrix.
Table 2: Conditional GC Results (p-values) for a Tri-Species System
| Causal Direction | Bivariate GC p-value | Conditional GC p-value (given 3rd species) | Inference |
|---|---|---|---|
| Sp. A → Sp. B | 0.003 | 0.450 | Indirect influence, mediated by Sp. C |
| Sp. A → Sp. C | 0.021 | 0.015 | Direct causal influence |
| Sp. B → Sp. C | 0.150 | 0.134 | No significant influence |
| Sp. C → Butyrate | <0.001 | <0.001 | Direct causal influence |
Protocol 2.3: Network Inference and Stability Validation
Objective: Construct a directed interaction network and assess its robustness. Procedure:
- Adjacency Matrix: Populate a matrix M where M[i,j] = 1 if GC test for j → i is significant (p < adjusted threshold), else 0.
- Bootstrapping: Generate N (e.g., 1000) surrogate datasets via block bootstrapping. Re-run conditional GC on each.
- Edge Confidence: Calculate the frequency (%) each directed edge appears across bootstrap runs. Retain edges with >70% confidence.
- Network Metrics: Compute stability-relevant metrics like modularity, reciprocity, and in/out degree distributions.
Title: Direct and Indirect Causal Links in a Tri-Species System
Application: Simulating Pharmacological Perturbation in a Microbiome Network
Objective: Use GC networks to predict systemic effects of a targeted antibacterial agent.
Experimental Protocol:
- Baseline Monitoring: For a model community (in vitro or in vivo), collect high-frequency abundance data for T_b time points to infer baseline GC network.
- Perturbation: Introduce a narrow-spectrum compound targeting a keystone pathogen (Node K).
- Post-Perturbation Monitoring: Continue monitoring for T_p time points.
- Dynamic GC Analysis: Employ sliding-window GC to compute time-varying networks.
- Impact Quantification:
- Calculate the shift in out-degree (influence) of Node K.
- Identify nodes with significant change in inbound GC influence (p < 0.05, bootstrap test).
- Track alterations in global metrics like network reciprocity.
Table 3: Simulated Network Metrics Pre- and Post-Perturbation
| Network Metric | Pre-Perturbation (Mean ± SE) | Post-Perturbation (Mean ± SE) | p-value (Change) |
|---|---|---|---|
| Edge Density | 0.32 ± 0.02 | 0.28 ± 0.03 | 0.12 |
| Reciprocity | 0.41 ± 0.05 | 0.22 ± 0.04 | 0.006 |
| Out-Degree of Target K | 4.1 ± 0.3 | 1.2 ± 0.5 | <0.001 |
| Modularity (Q) | 0.15 ± 0.02 | 0.31 ± 0.03 | 0.001 |
Title: Network Re-wiring Following Targeted Keystone Species Perturbation
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Materials for GC-Based Ecological Network Research
| Item | Function & Relevance | Example/Supplier |
|---|---|---|
| High-Throughput Sequencer | Generates species abundance time-series via 16S rRNA/ITS amplicon or metagenomic sequencing. Essential for variable definition. | Illumina NovaSeq, PacBio Sequel II |
| LC-MS/MS System | Quantifies metabolite concentrations (e.g., short-chain fatty acids, signaling molecules) for multi-modal GC analysis. | Thermo Fisher Orbitrap, Agilent Q-TOF |
| Anaerobic Chamber | Maintains strict conditions for in vitro cultivation of obligate anaerobic microbiota, enabling controlled perturbation studies. | Coy Laboratory Products, Baker Ruskinn |
| Time-Series Database | Specialized platform for storing, curating, and sharing longitudinal ecological data. | EcoTimeDB, pandas (Python), tsbox (R) |
| Granger Causality Software | Implements VAR modeling, conditional GC tests, and bootstrapping for robust network inference. | statsmodels.tsa.stattools.grangercausalitytests (Python), lmtest::grangertest() (R), MVGC Toolbox (MATLAB) |
| Network Visualization & Analysis Suite | Computes graph metrics, performs community detection, and visualizes directed networks. | Cytoscape, networkx (Python), igraph (R/Python) |
Why Ecological Networks? The Need for Causal Inference in Microbiome and Multi-Omics Data.
High-throughput sequencing and mass spectrometry generate vast multi-omics datasets (16S rRNA, metagenomics, metatranscriptomics, metabolomics) detailing microbial community composition and function. Standard analytical methods, such as correlation-based network inference (e.g., SparCC, SPIEC-EASI), identify statistical associations but fail to distinguish true ecological interactions (e.g., competition, cross-feeding) from spurious correlations induced by confounding factors. This gap critically limits the translation of observational data into testable hypotheses for therapeutic intervention. Granger causality, a time-series-based method rooted in predictive precedence, provides a formal framework for inferring directed, potentially causal relationships, making it essential for constructing predictive ecological network models.
Quantitative Evidence: Correlation vs. Causation in Microbial Studies
Table 1: Comparative Performance of Network Inference Methods on Benchmark Microbial Time-Series Data
| Method Type | Example Algorithm | Key Principle | Average Precision (Recall) for Directed Edges | Major Limitation |
|---|---|---|---|---|
| Correlation | SparCC | Compositionally robust correlation | 0.22 (0.85) | Undirected; high false-positive rate for direct interactions |
| Conditional Independence | SPIEC-EASI | Graphical model inference via partial correlations | 0.31 (0.65) | Undirected; struggles with nonlinear effects |
| Information Theory | Transfer Entropy | Non-parametric information flow | 0.45 (0.55) | Computationally intensive; requires large sample size |
| Granger Causality | Vector Autoregressive (VAR)-based Granger | Predictive precedence in time | 0.68 (0.60) | Requires dense temporal sampling; linear assumptions |
| Nonlinear Granger | Kernel or Random Forest-based | Nonlinear predictive precedence | 0.72 (0.58) | Very high computational demand; risk of overfitting |
Data synthesized from recent benchmarking studies (e.g., Faust et al., 2022; Tipton et al., 2023) using simulated microbial communities with known ground-truth interactions.
Core Protocol: Granger Causality Ecological Network Inference from Multi-Omics Time-Series
Protocol Title: Inferring Directed Microbial Interaction Networks Using Vector Autoregressive Granger Causality on Integrated Multi-Omics Time-Series Data.
Objective: To construct a directed, potentially causal ecological network from aligned 16S rRNA (relative abundance) and metabolomics (peak intensity) time-series data.
Materials & Preprocessing:
- Input Data: Matrices of OTU/ASV relative abundances (
X_abun) and metabolomic feature intensities (X_metab) acrossntime points formsamples. - Normalization: CLR-transform
X_abun. StandardizeX_metab(z-score per feature). - Lag Specification: Determine optimal time lag (
k) using AIC/BIC criteria on preliminary VAR models.
Step-by-Step Workflow:
- Data Integration & Lagging: Create a combined data matrix
[X_abun, X_metab]. Generate lagged matrices for lags1tok. - VAR Model Fitting: For each variable
Y_i(e.g., a specific microbe or metabolite), fit two models:- RESTRICTED Model (R):
Y_i(t) = f( past of Y_i ). - FULL Model (F):
Y_i(t) = f( past of Y_i, past of X_j ), whereX_jis a potential causal driver.
- RESTRICTED Model (R):
- Granger Causality Test: Perform an F-test comparing the residual sum of squares (RSS) of models
RandF.F-statistic = [(RSS_R - RSS_F) / k] / [RSS_F / (n - 2k - 1)].- The p-value indicates if
X_jGranger-causesY_i.
- Multiple Testing Correction: Apply False Discovery Rate (FDR, e.g., Benjamini-Hochberg) correction across all tested variable pairs.
- Network Visualization: Construct a directed adjacency matrix where a significant link (
FDR < 0.05) fromX_jtoY_iis represented as a directed edge. Visualize using force-directed layouts in Cytoscape or Gephi.
Diagram Title: Granger Causality Inference Protocol Workflow
Signaling Pathway Example: Inferring a Bile Acid-Mediated Host-Microbe Axis
Granger causality can disentangle complex host-microbe-metabolite pathways. A hypothesized causal chain is: Microbe A → Bile Acid Metabolite X → Host Gene Y.
Diagram Title: Inferred Bile Acid Mediated Host-Microbe Causal Chain
The Scientist's Toolkit: Key Reagent Solutions for Validation
Table 2: Essential Research Reagents for Validating Causal Microbiome Interactions
| Reagent / Material | Provider Examples | Function in Causal Inference |
|---|---|---|
| Gnotobiotic Mouse Models | Taconic, Jackson Labs | Provides a sterile (germ-free) host to colonize with defined microbial consortia for direct testing of inferred causal relationships. |
| Defined Microbial Consortia | ATCC, BEI Resources | Enables reconstruction of synthetic communities based on network nodes for targeted perturbation experiments. |
| Stable Isotope-Labeled Substrates (¹³C, ¹⁵N) | Cambridge Isotopes, Sigma-Aldrich | Traces metabolic flux from a donor microbe to a recipient, providing direct evidence of cross-feeding predicted by causal links. |
| Bile Acid Receptor Agonists/Antagonists (e.g., GW4064, Z-guggulsterone) | Tocris, Cayman Chemical | Pharmacologically manipulates specific host pathways (e.g., FXR) to test the causal role of metabolite-mediated host responses. |
| CRISPRi/a Systems for Gut Bacteria | Custom synthesis (e.g., IDT) | Enables targeted knockdown/activation of specific bacterial genes in situ to validate their causal role in community dynamics. |
| Anaerobic Culture Media (e.g., YCFA, BHI) | Anaerobe Systems, HiMedia | Supports the cultivation of fastidious anaerobic gut species for in vitro validation of pairwise interactions. |
Granger causality (GC) provides a operational, data-driven definition of causation for time series data. Within ecological interaction networks and drug development, it is used to infer directional influences (e.g., Species A precedes and predicts changes in Species B; Drug target modulation precedes disease marker change). A core philosophical consideration is that GC identifies predictive causality ("X Granger-causes Y if past values of X contain information that helps predict Y beyond the information contained in past values of Y alone"), not necessarily mechanistic causality. In dynamic biological systems, correlations can arise from common drivers, feedback loops, or indirect pathways, making the distinction critical.
Key Assumptions and Their Implications
| Assumption | Implication in Ecological/Drug Networks | Violation Consequence |
|---|---|---|
| Temporality Cause must precede effect. | Allows inference of directionality in species interactions or signaling pathways. | Leads to spurious causality if measurement lags are misaligned or systems are faster than sampling. |
| Causal Sufficiency All relevant confounding variables are included in the model. | Omitting a key species or cellular component can reverse or obscure true causal links. | Inferred GC network is incomplete or incorrect (e.g., missing hidden common regulator). |
| Stationarity The underlying data-generating process is stable over time. | Critical for translating in vitro findings to in vivo models or across treatment phases. | Parameter estimates are unreliable; causal links may appear/disappear artificially. |
| Linearity The GC model captures linear interactions. | Simplifies computation but may miss threshold effects, saturation, or oscillatory dynamics. | Non-linear causal relationships remain undetected or are mischaracterized. |
| Method | Key Metric | Optimal Use Case | Computational Load | Sensitivity to Violations |
|---|---|---|---|---|
| Vector Autoregression (VAR) | F-statistic, p-value | Linear, stationary systems with moderate variable count. | Low to Moderate | High (to sufficiency, stationarity) |
| Transfer Entropy (TE) | Bits of information transfer | Non-linear systems, non-parametric analysis. | High (requires more data) | Moderate (to sufficiency) |
| Convergent Cross Mapping (CCM) | Cross-map skill (ρ) | Weakly to moderately coupled dynamic systems (e.g., predator-prey). | High | Low (to stationarity) |
| Partial Granger Causality | Conditional F-statistic | High-dimensional data with observed confounders. | Moderate | Reduces sensitivity to latent variables |
Application Notes & Protocols
Protocol: Preprocessing for GC Analysis in Microbial Time-Series
Aim: Prepare 16S rRNA or metagenomic sequencing time-series data for reliable GC inference.
- Normalization: Convert raw reads to relative abundance or use a centered log-ratio (CLR) transformation to address compositionality.
- Imputation & Filtering: Apply Kalman filtering or simple interpolation for minor missing data. Remove taxa present in <10% of time points.
- Stationarity Check: Apply the Augmented Dickey-Fuller test to each taxa series. Differencing (d=1) is applied to non-stationary series.
- Lag Selection: Use Akaike Information Criterion (AIC) or Bayesian Information Criterion (BIC) on a full VAR model to determine optimal lag (k). Maximum lag should not exceed 1/4 of time-series length.
- Embedding (for CCM): Use simplex projection to determine optimal embedding dimension (E) for state-space reconstruction.
Protocol: GC Inference forIn VitroSignaling Pathway Dissection
Aim: Determine causal ordering of phospho-protein activation post-stimulation.
- Experimental Data: Collect high-throughput (e.g., Luminex, phospho-flow) time-course data (t=0, 2, 5, 15, 30, 60 min) under control and perturbed (kinase inhibitor) conditions.
- Model Construction: For each condition, fit a multivariate VAR model: X(t) = Σᵢ AᵢX(t-i) + ε(t), where X is a vector of protein activity levels.
- Causality Testing: Perform block F-tests to determine if including past values of variable j significantly reduces the prediction error for variable i. Use false discovery rate (FDR) correction for multiple comparisons.
- Validation Perturbation: Statistically compare GC networks from control vs. inhibited conditions. A true causal link from protein A→B should diminish or disappear when A is inhibited.
Protocol: Validating GC Networks with Interventional Data
Aim: Ground statistical causality in mechanistic biology.
- Generate Hypotheses: From the inferred GC network, select top candidate causal links (e.g., "Inflammatory Cytokine A → Disease Marker B").
- Design Intervention: Use siRNA, CRISPRi, or a selective small-molecule inhibitor to knock down/out the putative cause (Cytokine A or its receptor).
- Measure Outcome: Quantify the target effect (Disease Marker B) over the same time frame used for GC analysis.
- Assessment: A significant reduction in the GC strength (or its significance) of the A→B link in the perturbed dataset, coupled with an experimental reduction in B, confirms a likely mechanistic causal link.
Visualizations
Title: Granger Causality Analysis and Validation Workflow
Title: Causal Structures in Dynamic Networks
The Scientist's Toolkit: Essential Reagents & Materials
| Item / Solution | Function in GC-Related Research | Example / Specification |
|---|---|---|
| High-Throughput Time-Course Assay Kits | Generate dense, multivariate time-series data for GC input. | Luminex multi-analyte panels, Phospho-kinase array kits, RT-qPCR panels. |
| Selective Pathway Inhibitors | Experimental validation of inferred causal links via perturbation. | Kinase inhibitors (e.g., SB203580 for p38 MAPK), receptor antagonists, siRNA pools. |
| Metagenomic Sequencing Reagents | Profile ecological network nodes (microbial taxa/genes) over time. | 16S rRNA gene primers (V4 region), shotgun library prep kits (Illumina). |
| Time-Lapse Live-Cell Imaging Reagents | Capture dynamic single-cell trajectories for causal analysis. | Fluorescent biosensors (FRET-based), vital dyes, photoactivatable proteins. |
| Statistical Software Packages | Perform GC calculations, network inference, and significance testing. | R: vars, lmtest, TransferEntropy; Python: statsmodels, pycausality. |
Implementing Granger Causality: Step-by-Step Methods for Biomedical Ecological Data
This protocol details the experimental and computational prerequisites for generating high-resolution time-series data to infer Granger-causal ecological interaction networks within host-associated microbial communities. Within the broader thesis on "Granger Causality Ecological Interaction Networks Research," this framework is foundational. It enables the distinction between correlation and temporal precedence, a core requirement for hypothesizing driver-response relationships between microbial taxa and host molecules (e.g., metabolites, cytokines) in dynamic systems like the gut.
Core Data Prerequisites: Specifications & Rationale
The validity of Granger causality analysis is contingent on specific data characteristics.
Table 1: Minimum Data Specifications for Time-Series Granger Causality Analysis
| Parameter | Specification | Rationale |
|---|---|---|
| Temporal Resolution | Minimum of 10-15 time points per condition/individual. | Enables reliable estimation of lagged relationships and model convergence. |
| Sampling Frequency | Must be faster than the rate of change of the processes studied (e.g., hours for metabolites, days for community shifts). | Prevents aliasing and ensures causal signals are captured. |
| Replicate Strategy | Biological replicates (n ≥ 5 independent hosts/cohorts) with matched longitudinal sampling. | Controls for individual variation and establishes generalizability of inferred networks. |
| Data Types | Paired, matched samples: 16S rRNA/ITS-seq or shotgun metagenomics (microbial abundance) + Host molecular profiling (e.g., Metabolomics via LC-MS, Proteomics). | Provides the dual-variable input (X -> Y) required for pairwise or multivariate Granger tests. |
| Data Normalization | Required for both data types: Compositional (e.g., CLR for microbes) and Batch-effect correction. | Reduces false positives from spurious correlations inherent in compositional data. |
| Missing Data | <5% missingness per feature. Impute using methods like Kalman filtering for time-series. | Granger causality models require complete, aligned time-series vectors. |
Table 2: Example Sampling Schedule for Murine Gut Microbiome-Host Metabolome Study
| Time Point | Day | Relative to Perturbation | Key Measurements |
|---|---|---|---|
| T0 | 0 | Baseline (Pre-perturbation) | Fecal DNA, Cecal content (metabolomics), Serum. |
| T1 | 1 | Early Response | Fecal DNA, Cecal content. |
| T2 | 2 | Acute Phase | Fecal DNA, Cecal content, Serum. |
| T3 | 4 | Transition | Fecal DNA, Cecal content. |
| T4 | 7 | Early Stabilization | Fecal DNA, Cecal content, Serum. |
| T5 | 10 | New Steady State | Fecal DNA, Cecal content, Serum. |
Experimental Protocol: Longitudinal Sampling from a Murine Model
Protocol 2.1: Coordinated Fecal and Tissue Sampling for Multi-Omics
Objective: To collect matched, longitudinal samples for microbial genomic and host metabolomic profiling from individual mice before, during, and after a dietary or pharmacological perturbation.
Materials:
- Specific-pathogen-free (SPF) mice (C57BL/6J, 8-10 weeks old, n≥10).
- Metabolic cages (for individualized housing and fecal collection).
- Sterile forceps, surgical scissors.
- DNA/RNA Shield (Zymo Research) or similar nucleic acid stabilization buffer.
- Pre-chilled methanol:acetonitrile:water (40:40:20, v/v) for metabolite quenching/extraction.
- Cryogenic vials, labeled for individual and time point.
- Liquid nitrogen and -80°C freezer.
Procedure:
- Acclimatization: House mice individually in metabolic cages for 7 days with ad libitum access to standard chow and water.
- Baseline Sampling (T0): Collect fresh fecal pellets directly from cage floor using sterile forceps. Aliquot:
- For Genomics: Immediately place 1 pellet (~100 mg) into a tube containing 1 mL DNA/RNA Shield. Vortex, store at 4°C (short-term) or -20°C.
- For Metabolomics: Snap-freeze 2 pellets in liquid N₂, store at -80°C.
- Collect blood via submandibular bleed for serum isolation; snap-freeze serum in liquid N₂.
- Perturbation & Longitudinal Sampling: Administer defined perturbation (e.g., antibiotic in drinking water, high-fat diet). At each defined time point (T1...T5), repeat Step 2 for fecal collection.
- Terminal Time Point (T5): Euthanize mouse. Aseptically collect cecal content and distal ileum tissue. Snap-freeze all samples in liquid N₂. Store all samples at -80°C until processing.
Protocol 2.2: DNA Extraction and 16S rRNA Gene Amplicon Sequencing
Objective: To generate microbial community abundance profiles from stabilized fecal samples.
Procedure:
- Cell Lysis: Homogenize stabilized fecal sample. Use a bead-beating mechanical lysis step (e.g., with 0.1 mm glass beads) for 5 min.
- DNA Extraction: Perform extraction using a dedicated stool DNA kit (e.g., QIAamp PowerFecal Pro DNA Kit) following manufacturer's instructions, including optional RNase A step.
- Amplification: Amplify the V4 hypervariable region of the 16S rRNA gene using dual-indexed primers (515F/806R). Use a high-fidelity polymerase and minimum PCR cycles (25-30).
- Sequencing: Pool purified amplicons in equimolar ratios. Sequence on Illumina MiSeq platform with 2x250 bp paired-end chemistry.
Data Preprocessing & Granger Causality Analysis Workflow
Preprocessing for Causal Inference
Key Signaling Pathways in Microbiome-Host Interaction
Microbial Metabolites Activate Host Pathways
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 3: Research Reagent Solutions for Time-Series Microbiome-Host Studies
| Item | Function / Rationale | Example Product |
|---|---|---|
| Nucleic Acid Stabilizer | Preserves microbial community structure at moment of sampling, prevents shifts. Critical for accuracy. | Zymo Research DNA/RNA Shield |
| Mechanical Lysis Beads | Ensures complete lysis of diverse microbial cell walls (Gram+, Gram-, spores) for unbiased DNA yield. | 0.1 mm & 0.5 mm Zirconia/Silica Beads |
| Stool DNA Extraction Kit | Optimized for inhibitor removal from complex matrices; yields PCR-ready microbial DNA. | QIAGEN QIAamp PowerFecal Pro DNA Kit |
| PCR Inhibitor Removal Beads | Further cleans DNA post-extraction for optimal library prep efficiency. | MagBio HighPrep PCR Clean-up |
| 16S rRNA Primers (V4) | Standardized, barcoded primers for reproducible amplification of the target region. | Illumina 515F/806R Primer Set |
| Metabolite Quenching Solution | Instantaneously halts enzymatic activity to capture in vivo metabolite levels. | Cold Methanol:ACN:Water (40:40:20) |
| Internal Standards (Metabolomics) | Enables quantitative and QC analysis across samples/runs for LC-MS data. | Cambridge Isotope Laboratories MSK-ISTD Kit |
| Cytokine Multiplex Assay | Measures concurrent host inflammatory response from limited sample volume (e.g., serum). | Luminex xMAP Technology Assays |
| VAR Model Software Package | Implements Granger causality and network inference on time-series data. | R vars or granger package |
This protocol details the application of Vector Autoregression (VAR) models and the associated F-test for Granger causality, a cornerstone methodology in modern ecological interaction network research. Within the broader thesis on "Inferring Trophic and Non-Trophic Interactions in Complex Ecosystems," VAR modeling provides a statistical framework to move beyond correlation and assess potential predictive, causal-like relationships between time-series variables, such as species abundances, nutrient levels, or environmental parameters. This approach is critical for generating testable hypotheses about ecosystem dynamics, stability, and response to perturbations, with direct relevance for conservation biology, natural resource management, and understanding the ecological impacts of pharmaceutical compounds.
Core Theoretical Framework
A Vector Autoregression (VAR) model of order p (VAR(p)) for a k-dimensional time series vector Yt = (y1t, y2t, ..., ykt)' is defined as:
Yt = c + Φ1Yt-1 + Φ2Yt-2 + ... + ΦpYt-p + εt
where c is a vector of constants, Φi are *k×k* coefficient matrices, and εt is a vector of white noise error terms.
Granger Causality Testing via F-test: A variable x is said to "Granger-cause" variable y if past values of x contain statistically significant information for predicting y, above and beyond the information contained in past values of y itself. This is tested by comparing two models:
- Restricted Model: y_t regressed on p lags of y.
- Unrestricted Model: y_t regressed on p lags of both y and x.
The test statistic is an F-test comparing the Residual Sum of Squares (RSS) of the two models.
Experimental Protocol: Implementing VAR & Granger Causality for Ecological Time-Series
Protocol 3.1: Data Preprocessing & Model Specification
Objective: Prepare multivariate ecological time-series data for VAR modeling. Steps:
- Data Collection: Assemble k time-series variables (e.g., log-transformed abundance of species A, B, C; daily temperature; nutrient concentration). Ensure equal time intervals and length (T observations).
- Stationarity Testing: Test each variable for unit roots using the Augmented Dickey-Fuller (ADF) test. Non-stationary series must be differenced until stationarity is achieved. This is critical to avoid spurious regression.
- Lag Length Selection: Using the stationary data, determine the optimal lag order p using information criteria (AIC, BIC, HQIC) computed from VAR models of increasing lag order. The lag order minimizing the criteria is selected.
Protocol 3.2: Model Estimation & Diagnostic Checking
Objective: Fit the VAR(p) model and validate its statistical assumptions. Steps:
- Estimation: Estimate the parameters (c, Φ1,..., Φp) of the VAR(p) model using Ordinary Least Squares (OLS).
- Residual Diagnostics: Test the model residuals for:
- Serial Correlation: Use Portmanteau test (e.g., Ljung-Box) on residual series.
- Normality: Use Jarque-Bera test.
- Heteroskedasticity: Use ARCH-LM test.
- Stability: Ensure all roots of the characteristic polynomial lie inside the unit circle.
- Model Refinement: If diagnostics fail, consider adding lags, transforming variables, or including deterministic trends/seasonal dummies.
Protocol 3.3: Granger Causality Significance Testing (F-test)
Objective: Formally test for pairwise Granger causal relationships within the ecosystem network. Steps:
- Hypothesis Formulation: For each ordered pair of variables (x, y), state:
- H₀: x does not Granger-cause y (all coefficients on lagged x in the y equation are zero).
- H₁: x does Granger-cause y (at least one coefficient is non-zero).
- Compute Restricted & Unrestricted RSS:
- Estimate the restricted equation for y (lags of y only). Obtain RSSR.
- Estimate the unrestricted equation for y (lags of y and x). Obtain RSSUR.
- Calculate F-statistic:
- F = [(RSSR - RSSUR) / p] / [RSS_UR / (T - 2p - 1)]
- where p is the lag order, T is sample size.
- Significance Assessment: Compare the calculated F-statistic to the critical value from the F-distribution with (p, T - 2p - 1) degrees of freedom. Reject H₀ if F > F_critical (p-value < α, typically 0.05).
Data Presentation & Results Interpretation
Table 1: Optimal Lag Order Selection for VAR Model (Example: Phytoplankton-Zooplankton-Nutrients System)
| Lag | Akaike Information Criterion (AIC) | Schwarz Criterion (BIC) | Hannan-Quinn Criterion (HQIC) |
|---|---|---|---|
| 0 | 15.234 | 15.345 | 15.276 |
| 1 | 8.456* | 8.901* | 8.623* |
| 2 | 8.521 | 9.301 | 8.812 |
| 3 | 8.603 | 9.717 | 9.018 |
*Indicates selected lag order. Conclusion: p=1 is chosen based on all three criteria.
Table 2: Pairwise Granger Causality F-Test Results (p=1, α=0.05)
| Null Hypothesis (H₀) | F-Statistic | P-Value | Conclusion (α=0.05) |
|---|---|---|---|
| Zooplankton ⇏ Phytoplankton | 6.724 | 0.012 | Reject H₀ (Causal Link) |
| Phytoplankton ⇏ Zooplankton | 1.205 | 0.277 | Fail to Reject H₀ |
| Phosphate ⇏ Phytoplankton | 9.832 | 0.003 | Reject H₀ (Causal Link) |
| Phytoplankton ⇏ Phosphate | 0.873 | 0.354 | Fail to Reject H₀ |
| Phosphate ⇏ Zooplankton | 2.457 | 0.123 | Fail to Reject H₀ |
| Zooplankton ⇏ Phosphate | 1.099 | 0.299 | Fail to Reject H₀ |
Interpretation: The analysis suggests a unidirectional Granger-causal network: Phosphate → Phytoplankton → Zooplankton. Past phosphate levels help predict phytoplankton, and past phytoplankton help predict zooplankton, but not vice-versa, aligning with a bottom-up trophic control hypothesis.
Visualizations
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents & Software for VAR/Granger Causality Analysis
| Item Name / Category | Specific Example / Platform | Function in Analysis |
|---|---|---|
| Statistical Software | R (vars, lmtest, urca packages), Python (statsmodels), Stata, EViews |
Provides functions for unit root testing, VAR estimation, lag selection, and F-test computation. |
| Data Curation Tool | R (tidyverse), Python (pandas), MATLAB |
Enables cleaning, transformation (log/difference), and structuring of multivariate time-series data. |
| Stationarity Test | Augmented Dickey-Fuller (ADF) Test, KPSS Test | Diagnoses unit root non-stationarity, guiding necessary data transformations. |
| Lag Selection Criterion | Akaike (AIC), Bayesian (BIC), Hannan-Quinn (HQIC) | Objectively determines the optimal number of lags (p) for the VAR model. |
| Diagnostic Test Suite | Portmanteau (Liung-Box) test, ARCH-LM test, Jarque-Bera test | Validates model adequacy by testing residuals for serial correlation, heteroskedasticity, and normality. |
| Visualization Package | R (ggplot2, DiagrammeR), Python (matplotlib, graphviz) |
Creates publication-quality graphs of time-series, network diagrams, and workflow charts. |
| High-Performance Computing (HPC) | University cluster, Cloud computing (AWS, GCP) | Facilitates analysis of large ecological datasets (many species/variables over long time periods). |
This document provides application notes and protocols for the application of Regularized Vector Autoregression (LASSO-VAR) and Sparse Graphical Models, key methodological pillars for inferring Granger-causal ecological interaction networks from high-dimensional, short-panel time-series data. The broader thesis investigates species interaction dynamics (e.g., microbial communities, predator-prey systems) to model perturbation responses, with direct analogies to host-pathogen dynamics and drug mechanism-of-action analysis in development.
Core Methodological Framework
LASSO-VAR Model Specification
For an N-variate ecological time series Yt = (y1,t, ..., yN,t)´, the VAR(p) model is: Yt = A1Yt-1 + A2Yt-2 + ... + ApYt-p + εt where Ak are N×N coefficient matrices and εt ~ N(0, Σ). In high-dimensional settings (N > T), the LASSO-VAR imposes an L1 penalty to induce sparsity: min{A} Σt ||Yt - Σk=1p AkYt-k||22 + λ Σi,j,k |aij(k)| where λ is the regularization parameter controlling sparsity. A non-zero aij(k) indicates Granger causality from variable j to variable i at lag k.
Sparse Graphical Model for Innovation Structure
The residual precision matrix Ω = Σ-1 is estimated via the Graphical LASSO (GLASSO): maxΩ ≻ 0 log det(Ω) - tr(SΩ) - ρ ||Ω||1 where S is the sample covariance matrix of VAR residuals, and ρ is the L1 penalty. A non-zero ωij in Ω indicates a conditional dependence (partial correlation) between variables i and j, after accounting for all lagged temporal effects, representing contemporaneous ecological interactions.
Experimental Protocols
Protocol A: Network Inference from Time-Series Abundance Data
Objective: Infer a directed (Granger-causal) and undirected (contemporaneous) ecological interaction network from high-dimensional species abundance time-series.
Input: T×N matrix of normalized abundance counts (e.g., 16S rRNA, metagenomic, or population survey data) across N species/OTUs over T time points.
Preprocessing:
- Normalization: Apply a centered log-ratio (CLR) transformation or variance-stabilizing transformation to compositional data.
- Stationarity: Apply first-differencing if needed (confirmed via Augmented Dickey-Fuller test).
- Lag Selection: Determine maximum lag p using information criteria (AIC/BIC) on a small-scale unrestricted VAR.
LASSO-VAR Estimation (using glmnet or BigVAR in R):
- Vectorize: Rearrange the VAR into a multivariate regression.
- Tune λ: Perform 10-fold time-series cross-validation (blocked CV) over a log-spaced λ grid (e.g., 100 values). Select λ that minimizes forecast MSE.
- Estimate: Fit the model with the optimal λ.
- Extract Network: Construct adjacency matrix G where Gij = 1 if Σk=1p |aij(k)| > 0 (threshold: 1e-4).
GLASSO on Residuals:
- Compute Residuals: Êt = Yt - Ŷt from the LASSO-VAR model.
- Tune ρ: Use 10-fold CV on the residual matrix Ê to select ρ maximizing the likelihood.
- Estimate Ω: Fit the GLASSO model (
glassoR package). - Extract Contemporaneous Network: Construct adjacency matrix C where Cij = 1 if |ωij| > 0 (threshold: 1e-4).
Validation (Stability Selection):
- Subsample: Re-run the entire inference pipeline on 100 random subsamples (80% of time points).
- Calculate Edge Probabilities: An edge is considered stable if it appears in >80% of subsampled networks.
Protocol B: Perturbation Response Prediction & Drug Analogy Testing
Objective: Predict community response to a targeted perturbation (e.g., species removal, antibiotic introduction) and identify key mediator species.
Input: Inferred LASSO-VAR model, pre-perturbation time-series data, perturbation target (species j).
Procedure:
- Simulate Perturbation: Set all coefficients a•j(k) (effects from species j) to zero in the estimated A matrices.
- Dynamic Forecast: Using the last p observed time points as initial conditions, simulate the perturbed system forward for H time steps (post-perturbation horizon).
- Compare to Baseline: Simulate an unperturbed forecast from the same initial conditions.
- Identify Key Responders: Species i with the largest absolute divergence between perturbed and baseline trajectories are primary responders.
- Pathway Analysis: Use the combined G and C networks to trace all shortest paths from the perturbation target j to key responder i; these are candidate mechanistic pathways.
Validation via In Silico Knockouts:
- Compare predicted responder status against independent experimental data from gnotobiotic models or chemostat studies with targeted antimicrobials.
Table 1: Comparison of Regularization Methods for Ecological VAR
| Method | Penalty | Key Hyperparameter(s) | Optimal For | Computational Complexity | Sparsity Control | ||||
|---|---|---|---|---|---|---|---|---|---|
| LASSO-VAR | L1 ( | A | 1) | λ (regularization strength) | General-purpose, small-to-medium N | O(N²pT) | Global, uniform | ||
| Lag-Adaptive LASSO-VAR | Weighted L1 | λ, decay parameter | Prioritizing shorter lags | O(N²pT) | Lag-specific | ||||
| Hierarchical VAR | Group L1/L2 | λ, α (mixing) | Grouping species by taxonomy/function | O(N²pT * groups) | Structured sparsity | ||||
| Bayesian VAR | Hierarchical Shrinkage | Prior scales | Incorporating prior knowledge | High (MCMC) | Probabilistic |
Table 2: Typical Hyperparameter Ranges for Microbial Time-Series (N=50-200, T=50-500)
| Parameter | Description | Recommended Search Range | Tuning Method | Notes |
|---|---|---|---|---|
| VAR Lag (p) | Maximum temporal order | 1 to 5 | BIC on small VAR | Ecological processes often have short lags. |
| λ (LASSO-VAR) | Coefficient sparsity | Log-spaced: 10⁻⁴ to 10¹ | Time-series Blocked CV | Larger λ for smaller T/N ratio. |
| ρ (GLASSO) | Precision matrix sparsity | Log-spaced: 10⁻² to 1 | Standard 10-fold CV on residuals | |
| Stability Threshold | Edge selection probability | 0.7 to 0.9 | Stability Selection | Higher threshold reduces false positives. |
Visualization of Methodological Workflows
Title: LASSO-VAR and GLASSO Network Inference Workflow
Title: In Silico Perturbation Prediction Protocol
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Computational Tools & Resources
| Item / Software Package | Function in Analysis | Critical Parameters/Notes |
|---|---|---|
BigVAR (R package) |
Efficient estimation of LASSO-VAR models on high-dimensional data. | Implements multiple penalties (Basic, Lag, Hierarchical). Use cv.BigVAR() for tuning. |
glmnet (R package) |
Core engine for LASSO regression. Used for custom VAR vectorization. | Family=mgaussian for multivariate. Ensure standardize=FALSE if data is preprocessed. |
glasso / huge (R packages) |
Estimates sparse precision matrix (GLASSO) from residuals. | huge provides faster approximate methods and rich model selection. |
igraph / networkD3 (R packages) |
Visualization and analysis of inferred ecological networks. | Calculate centrality measures (betweenness) to identify keystone species. |
| Stability Selection Script | Custom R/Python script for subsampling & edge probability calculation. | 100 subsamples at 80% sampling ratio is a robust default. |
| Normalized Abundance Data | Preprocessed, CLR-transformed species count matrix. | Essential to account for compositionality. Use compositions::clr(). |
| High-Performance Computing (HPC) Cluster | For cross-validation and stability selection on N>100. | Parallelize over λ/ρ grid and subsamples. |
| Reference Ecological Networks | In vitro or gnotobiotic model data for validation. | e.g., defined microbial community (SynCom) time-series post-antibiotic. |
This document provides detailed application notes and protocols for advanced Granger causality methods, framed within a broader thesis investigating ecological interaction networks and their perturbation. The nonlinear and high-dimensional nature of species interdependencies, gene regulatory networks, and host-pathogen-drug interactions in ecology and pharmacology demands moving beyond traditional linear vector autoregression. This work details the implementation of Kernel Granger Causality (KGC) and related nonparametric techniques to infer directed influence in complex systems, with direct applications in elucidating drug mechanisms and ecological resilience.
Theoretical Framework and Key Concepts
Kernel Granger Causality (KGC): A nonlinear extension of Granger causality that operates in reproducing kernel Hilbert spaces (RKHS). By mapping time-series data into a high-dimensional feature space via a kernel function (e.g., Gaussian, polynomial), KGC can detect nonlinear causal relationships. The core test involves comparing the prediction error of a future value (Y(t+1)) using the history of (Y) alone versus using the histories of both (Y) and (X).
Nonparametric Approaches: Include conditional mutual information-based methods, transfer entropy, and local process approximations. These models make minimal assumptions about the underlying functional form of interactions.
Application Notes: Key Experimental Scenarios
Analyzing Pharmacodynamic Interactions in Drug Combinations
Objective: To decipher synergistic or antagonistic causal pathways between two drugs (A & B) on a cellular outcome (e.g., apoptosis rate) over time. Rationale: Linear GC may miss nonlinear saturation or feedback effects. KGC can reveal how the temporal dynamics of Drug A's pathway causally influence the dynamics of Drug B's target pathway.
Unraveling Trophic Cascades in Microbial Ecosystems
Objective: To infer causal links in microbiome time-series data post-antibiotic perturbation. Rationale: Species interactions are often nonlinear (e.g., logistic growth, allelopathy). Nonparametric GC can identify keystone species and directional influences in recovery dynamics.
Detailed Experimental Protocols
Protocol 4.1: Kernel Granger Causality Analysis for High-Throughput Time-Series Data
I. Experimental Design & Data Acquisition
- System Perturbation: Design interventions (e.g., drug pulse, species removal) to generate informative dynamics. Include replicate time series (n ≥ 5).
- High-Frequency Monitoring: Collect equidistant temporal samples. The sampling rate must capture the hypothesized interaction timescale (e.g., minutes for phosphorylation, hours for population growth).
- Data Preprocessing: Detrend, normalize, and handle missing values. For microbial data, convert sequencing counts to relative abundances or use appropriate transformations.
II. Computational Analysis Workflow
- Embedding & Lag Selection:
- Use false nearest-neighbors or mutual information to determine optimal embedding dimension (m) and time lag (τ) for state-space reconstruction.
- Kernel Function Selection:
- Gaussian (RBF) Kernel: Default choice for capturing general smooth nonlinearities. Use cross-validation to select the bandwidth parameter (σ).
- Polynomial Kernel: Suitable for suspected power-law relationships.
- Causality Testing Implementation:
- Let ({xt, yt}, t=1,...,T) be two stationary time series.
- Formulate the following regularized least-squares problems in the RKHS:
- Restricted Model: Predict (yt) using its own history (Y^{past}t = [y{t-1}, ..., y{t-m}]).
- Full Model: Predict (yt) using (Y^{past}t) and (X^{past}t = [x{t-1}, ..., x{t-m}]).
- Compute the prediction errors: (\epsilonR) and (\epsilonF).
- The KGC statistic from (X \rightarrow Y) is: (F{X\rightarrow Y} = \ln(\frac{\epsilonR}{\epsilonF})). A significance test is performed via a permutation bootstrap (Protocol 4.2).
III. Validation & Controls
- Surrogate Data Test: Generate phase-randomized or iterative-amplitude-adjusted surrogate time series for (X) to destroy any potential causal link. The computed KGC on surrogate data should be non-significant.
- Conditional Analysis: To avoid indirect causation, condition on potential confounder series (Z) by including (Z^{past}_t) in both models.
Protocol 4.2: Permutation Bootstrap for Significance Testing of KGC
- Compute the true KGC statistic (F{obs}) for the original paired series ({xt, y_t}).
- For b = 1 to B (B = 1000-5000) do:
- Randomly shuffle the time indices of the presumed cause series (xt) to create (x^t), destroying its temporal order and any causal link to (y_t) while preserving its marginal distribution.
- Compute the KGC statistic (Fb) for the pair ({x^t, y_t}).
- Construct an empirical null distribution from ({F1, ..., FB}).
- Calculate the p-value: (p = \frac{#{Fb \geq F{obs}} + 1}{B + 1}).
- Reject the null hypothesis (no causality) if (p < \alpha) (e.g., 0.05), corrected for multiple comparisons.
Protocol 4.3: Transfer Entropy Estimation for Discrete/Categorical Signals
Applicability: For neural spike trains, discrete behavioral states, or binarized gene expression.
- Symbolic Encoding: Convert continuous signals to discrete states using appropriate binning.
- Estimation:
- Transfer Entropy from X to Y: (TE{X\rightarrow Y} = \sum p(y{t+1}, y^{(k)}t, x^{(l)}t) \log \frac{p(y{t+1} | y^{(k)}t, x^{(l)}t)}{p(y{t+1} | y^{(k)}_t)})
- where (y^{(k)}t, x^{(l)}t) are the k- and l-dimensional past states.
- Use a biased-corrected estimator (e.g., Effective Transfer Entropy) to account for finite sample effects.
Data Presentation: Comparative Analysis of GC Methods
Table 1: Comparison of Granger Causality Methodologies for Complex Interactions
| Feature | Linear Vector Autoregression (VAR) | Kernel Granger Causality (KGC) | Transfer Entropy (TE) |
|---|---|---|---|
| Core Assumption | Linear interactions, Gaussian residuals | Nonlinear interactions reproducible via kernel | General statistical dependency (non-parametric) |
| Model Specification | Lag order (p) | Kernel type & parameters (e.g., σ), Lag (m) | Embedding dimensions (k, l), binning strategy |
| Data Requirements | Stationary continuous series | Stationary series, larger sample size needed | Large sample size for reliable PDF estimation |
| Strength | Fast, interpretable coefficients | Captures complex nonlinearities, strong theory | Model-free, applicable to any type of data |
| Weakness | Misses nonlinear causality | Computationally intensive, choice of kernel | High estimator variance, requires much data |
| Typical Use Case | Preliminary screening, linear systems | Pharmacodynamics, nonlinear ecosystems | Neural circuits, discrete state systems |
Table 2: Example KGC Analysis of Simulated Microbial Species Interaction
| Causal Direction (X → Y) | True Model Lag | KGC Statistic (F) | p-value (Permutation) | Result |
|---|---|---|---|---|
| SpeciesA → SpeciesB | 2 | 0.452 | 0.003 | Detected |
| SpeciesB → SpeciesA | - | 0.021 | 0.412 | Not Detected |
| SpeciesA → MetaboliteM | 1 | 0.891 | <0.001 | Detected |
| SpeciesC → SpeciesA | 3 | 0.205 | 0.021 | Detected |
Visualization of Methodologies
Title: Kernel Granger Causality Analysis Workflow
Title: Drug Interaction Causal Pathway Analysis
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Resources for Implementing Nonlinear Granger Causality
| Item/Category | Example/Specific Product | Function in Research |
|---|---|---|
| Time-Series Data Generation | Live-cell imaging systems (Incucyte), Biosensors (FRET-based), High-throughput sequencer (Illumina NovaSeq) | Generates high-frequency, multivariate temporal data for causal analysis. |
| Computational Environment | Python (SciPy, scikit-learn, PyTorch), R (kernlab, rEDM, BigVAR), Julia (DynamicalSystems.jl) | Provides libraries for kernel methods, state-space reconstruction, and statistical testing. |
| Specialized Software | MuTE (Matlab Transfer Entropy), Causality Toolbox (gc-kernel), PCMCI+ (Python) | Offers dedicated, validated implementations of nonlinear GC and related algorithms. |
| Kernel Functions | Radial Basis Function (RBF), Polynomial, Linear, Sigmoid (via libraries) | Defines the feature space mapping; choice critically impacts sensitivity to different nonlinearities. |
| Validation Reagents | Pathway-specific inhibitors/activators (e.g., kinase inhibitors), CRISPRi knock-down pools | Provides ground-truth perturbation for experimental validation of inferred causal links. |
| High-Performance Compute | Cloud computing (AWS, GCP) or local cluster with GPU acceleration | Handles the computational load of permutation tests on large datasets or many variable pairs. |
Application Notes
In ecological interaction networks research, distinguishing direct causality from spurious correlations induced by dense connectivity is a fundamental challenge. Conditional Granger Causality (CGC) provides a mathematical framework to address this by statistically testing whether the past of one time-series variable X contains information that helps predict another variable Y, over and above the information contained in the past of all other observed variables in the network. This is critical for inferring true trophic interactions, host-pathogen dynamics, or stressor-response pathways from multivariate ecological time-series data, such as population counts, gene expression levels, or environmental sensor readings.
Key Quantitative Findings in Network Inference
Table 1: Comparison of Causality Inference Methods in Simulated Dense Networks (n=20 nodes, mean degree=8)
| Method | True Positive Rate (Mean ± SD) | False Positive Rate (Mean ± SD) | Computational Time (sec, Mean ± SD) | Key Assumption |
|---|---|---|---|---|
| Pairwise Granger Causality | 0.89 ± 0.05 | 0.41 ± 0.08 | 2.3 ± 0.7 | Network sparsity |
| Conditional Granger Causality | 0.85 ± 0.06 | 0.09 ± 0.03 | 18.7 ± 4.2 | Sufficient embedding |
| Transfer Entropy | 0.82 ± 0.07 | 0.15 ± 0.05 | 124.5 ± 32.1 | Stationarity |
| Bayesian Network Inference | 0.76 ± 0.08 | 0.11 ± 0.04 | 65.8 ± 12.4 | Acyclicity |
Table 2: Impact of Signal-to-Noise Ratio (SNR) on CGC Detection Performance
| SNR (dB) | Detection Power for Direct Links | Detection Power for Indirect Links | Optimal Model Order (Lag) |
|---|---|---|---|
| 30 | 0.98 | 0.02 | 3 |
| 20 | 0.95 | 0.05 | 3 |
| 10 | 0.81 | 0.12 | 2 |
| 5 | 0.62 | 0.25 | 1 |
Experimental Protocols
Protocol 1: Preprocessing Multivariate Ecological Time-Series Data for CGC Analysis
Objective: To prepare raw, observed time-series data (e.g., species abundance, metabolite concentration) for robust CGC analysis.
- Data Collection & Alignment: Ensure all N variables are sampled simultaneously at a consistent frequency. For missing data points (≤5%), use multiple imputation (e.g., multivariate imputation by chained equations). For gaps >5%, consider segmenting the time series.
- Detrending & Stationarity: Apply a first-difference or linear detrending filter to each series. Test for stationarity using the Augmented Dickey-Fuller test (p<0.05). If non-stationary, apply appropriate transformations (e.g., log, square root) or use a cointegration framework.
- Normalization: Z-score normalize each variable (subtract mean, divide by standard deviation) to prevent magnitude-based biases in model fitting.
- Model Order Selection: Using the normalized, stationary multivariate dataset, determine the optimal lag p (model order) for the Vector Autoregressive (VAR) model. Use the Bayesian Information Criterion (BIC) or Akaike Information Criterion (AIC) across a range of lags (e.g., 1 to 10). The lag minimizing the criterion is selected.
- Surrogate Data Testing (Optional): To establish a significance threshold, generate phase-randomized surrogate datasets for each variable. CGC values from the original data must exceed the 95th percentile of the CGC distribution from surrogates.
Protocol 2: Performing Conditional Granger Causality Analysis
Objective: To compute the CGC from variable X to variable Y conditioned on a set of other variables Z.
- Fit the Full VAR Model: Using the preprocessed data and selected model order p, fit a VAR model including all N variables: Y(t), X(t), and the set of conditioning variables Z(t). Estimate the residual covariance matrix Σ_full.
- Fit the Restricted VAR Model: Fit a second VAR model omitting the past values of the putative cause X. This model includes Y(t) and Z(t) only. Estimate its residual covariance matrix Σ_restricted.
- Compute CGC Value: Calculate the CGC from X to Y given Z using the formula:
CGC_(X→Y|Z) = ln( |Σ*_restricted*| / |Σ*_full*| )where |·| denotes the determinant. This value is always non-negative. - Statistical Testing: Perform an F-test on the model residuals or a likelihood-ratio test to assess if the inclusion of X's past leads to a statistically significant reduction in prediction error for Y. Correct for multiple comparisons across all tested pairs using the False Discovery Rate (FDR, e.g., Benjamini-Hochberg procedure) at α=0.05.
- Network Visualization: Construct a directed graph where nodes are variables and a directed edge from X to Y is drawn if the CGC is statistically significant.
Protocol 3: Validating CGC Networks with Perturbation Experiments
Objective: To empirically validate inferred causal links in an ecological or laboratory setting.
- Target Selection: From the inferred CGC network, select 3-5 high-strength putative direct causal links for validation.
- Controlled Perturbation: For a selected causal variable (e.g., predator species, a specific signaling molecule), design a controlled perturbation. This could be a pulse (acute removal/addition) or a press (sustained change) intervention.
- High-Resolution Monitoring: Monitor the dynamics of the target effect variable(s) and key network neighbors at a temporal resolution finer than the model's inferred lag time.
- Causal Contrast: Compare the response trajectory in the perturbed system to an unperturbed control. A validated causal link is supported if the effect variable's dynamics significantly deviate from the control prediction in the direction anticipated by the CGC model, while conditioned neighbors do not show the primary response.
- Iterative Refinement: Feed the perturbation response data back into the CGC modeling pipeline to refine model order and network structure.
Visualizations
CGC Analysis Workflow
Direct vs Indirect Causality
The Scientist's Toolkit
Table 3: Essential Research Reagents & Solutions for CGC-Based Network Research
| Item | Function & Application in CGC Research |
|---|---|
| Multivariate Time-Series Dataset | The core input. High-temporal-resolution measurements of multiple interacting variables (e.g., species counts, gene expression, environmental factors). Requires N > 50 time points for stable VAR estimation. |
| Stationarity Testing Suite (ADF/KPSS) | Software or code (e.g., statsmodels in Python) to verify the constant statistical properties of time-series data, a core assumption of standard Granger causality. |
| Vector Autoregressive (VAR) Model Fitting Package | Computational library (e.g., vars in R, statsmodels.tsa.VAR in Python) to estimate the full and restricted multivariate linear models central to CGC calculation. |
| Information Criterion Script (BIC/AIC) | Routine for optimal model order (lag) selection, critical to avoid under-fitting or over-fitting the temporal dependencies. |
| Statistical Inference Toolkit | Functions for F-test, likelihood-ratio test, and False Discovery Rate (FDR) correction to assign significance to computed CGC values. |
| Network Visualization Software | Tool (e.g., Cytoscape, Gephi, or NetworkX/Graphviz in Python) to render the final directed causal graph from significant CGC links. |
| Phase Randomization Surrogate Algorithm | Code to generate null datasets for establishing significance thresholds, helping to reject spurious causalities from linear correlations. |
| High-Resolution Ecological Sensors / Sequencers | Hardware for data collection (e.g., autonomous environmental sensors, qPCR, RNA-seq) to generate the necessary dense, longitudinal data. |
This application note details methodologies for reconstructing directed, ecological interaction networks within the gut microbiome under disease conditions. It is framed within a broader thesis on Granger causality ecological interaction networks research, which posits that temporal precedence and predictive capacity can infer causal ecological interactions from longitudinal multi-omics data. This approach moves beyond correlation to model how microbial abundances and metabolic activities may dynamically influence one another and the host in states such as Inflammatory Bowel Disease (IBD), colorectal cancer, and metabolic syndrome.
Table 1: Representative Microbial Taxa with Altered Interaction Patterns in Disease States
| Disease State | Taxa with Increased Causal Outflow (Influencers) | Taxa with Increased Causal Inflow (Responders) | Key Metabolite Correlates | Citation (Year) |
|---|---|---|---|---|
| Crohn's Disease | Escherichia coli (AIEC pathotype), Ruminococcus gnavus | Faecalibacterium prausnitzii, Roseburia spp. | Reduced Butyrate, Succinate | Lloyd-Price et al., 2019 |
| Ulcerative Colitis | Bilophila wadsworthia, Klebsiella pneumoniae | Akkermansia muciniphila, Bacteroides spp. | Increased Secondary Bile Acids, Sulfide | Schirmer et al., 2022 |
| Colorectal Cancer | Fusobacterium nucleatum, Bacteroides fragilis (ETBF) | Clostridium butyricum, Lactobacillus spp. | Polyamines, IL-17, Toxin B | Wirbel et al., 2021 |
| Type 2 Diabetes | Blautia spp., Acidaminococcus | Prevotella copri, Bifidobacterium spp. | Branched-Chain Amino Acids, Imidazole Propionate | Ruuskanen et al., 2022 |
Table 2: Common Granger Causality Network Metrics in Health vs. Disease
| Network Metric | Healthy State (Mean) | Disease State (Mean) | Interpretation |
|---|---|---|---|
| Network Density | 0.15 - 0.25 | 0.30 - 0.45 | More total inferred interactions in dysbiosis. |
| Interaction Sign Ratio (Positive:Negative) | 70:30 | 40:60 | Shift towards inhibitory/predatory interactions. |
| Average Path Length | 2.1 - 2.8 | 1.5 - 2.0 | More direct, potentially destabilizing interactions. |
| Clustering Coefficient | 0.10 - 0.20 | 0.05 - 0.12 | Breakdown of cooperative guild structure. |
Experimental Protocols
Protocol 3.1: Longitudinal Sample Collection & Metagenomic Sequencing for Granger Analysis
Objective: Obtain high-temporal-resolution data for time-series causal inference.
- Cohort Design: Recruit matched patient cohorts (disease vs. healthy control). For IBD, a flare-remission longitudinal design is optimal.
- Sampling: Collect fecal samples at high frequency (e.g., twice weekly for 8-12 weeks). Use standardized DNA/RNA stabilization buffers immediately upon collection.
- DNA Extraction: Use a bead-beating mechanical lysis kit (e.g., MagAttract PowerMicrobiome Kit) optimized for Gram-positive bacteria.
- Sequencing: Perform whole-metagenome shotgun sequencing (Illumina NovaSeq, 150bp paired-end, >10 million reads/sample). Include metatranscriptomic sequencing for active community analysis.
- Bioinformatic Processing: Process reads through an established pipeline:
- Quality trimming (Trimmomatic).
- Host read subtraction (Bowtie2 vs. human genome).
- Taxonomic profiling (MetaPhlAn4 for species-level abundance).
- Functional profiling (HUMAnN3 for pathway abundance).
- Time-Series Table Generation: Compile a matrix where rows are time points and columns are normalized species/pathway abundances.
Protocol 3.2: Inferring Granger Causal Networks using PCMCI
Objective: Apply a robust causal discovery algorithm to infer directed interaction networks.
- Data Preprocessing: Impute missing data (e.g., Kalman filter). Apply a centered log-ratio (CLR) transformation to compositional abundance data. Optionally detrend.
- Algorithm Selection: Implement the PCMCI (Peter-Clark Momentary Conditional Independence) algorithm, which is robust to autocorrelation and common confounders.
- Parameter Setting:
- Maximum Time Lag (τ_max): Set based on biological knowledge (e.g., 2-3 sampling intervals).
- Conditional Independence Test: Use linear or non-parametric (Gaussian process) tests based on data distribution.
- Significance Level (α): Apply a false discovery rate (FDR) correction (q < 0.05) across all tests.
- Network Construction: For each significant link (Xt-τ → Yt), record the direction, lag (τ), and strength (partial correlation coefficient). Aggregate links into a directed, weighted adjacency matrix.
- Validation: Perform sensitivity analysis on τ_max and α. Compare networks from randomized time series to establish a false-positive baseline.
Protocol 3.3: Experimental Validation of Microbial Interactions viaEx VivoCulturing
Objective: Validate computationally predicted interactions (e.g., inhibition, cross-feeding).
- Strain Isolation: Isalate target bacterial species from patient samples using selective media and anaerobic culture chambers (80% N2, 10% CO2, 10% H2, 37°C).
- Conditioned Media Experiment: a. Grow the putative "influencer" strain to late-log phase in defined medium. b. Centrifuge (8000 x g, 10 min) and filter-sterilize (0.22 µm) supernatant to create conditioned medium. c. Resuspend the "responder" strain in fresh medium (control) and conditioned medium. d. Monitor responder growth kinetics (OD600) and endpoint metabolomics (GC-MS) for 24-48 hours.
- Co-culture Experiment: Co-culture predicted interacting pairs in a chemostat or batch system. Use strain-specific qPCR or fluorescent markers to track individual population dynamics over time, comparing to mono-culture controls.
- Statistical Analysis: Compare growth parameters (lag time, max rate) and metabolite profiles using t-tests or ANOVA to confirm predicted facilitative or inhibitory interactions.
Visualizations
Title: Workflow for Granger Causal Network Reconstruction
Title: Example IBD Interaction Network & Outcomes
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions for Microbiome Interaction Studies
| Item / Reagent | Function / Application | Example Product / Note |
|---|---|---|
| DNA/RNA Stabilization Buffer | Preserves nucleic acid integrity in fecal samples at room temperature for transport/storage, critical for accurate longitudinal profiling. | OMNIgene•GUT, Zymo Research DNA/RNA Shield |
| Bead-Beating Lysis Kit | Mechanical disruption of tough microbial cell walls (e.g., Gram-positives, spores) for unbiased DNA extraction. | MP Biomedicals FastDNA Spin Kit, Qiagen PowerSoil Pro Kit |
| Defined Minimal Medium | For ex vivo validation experiments, allows precise control of nutrients to study cross-feeding and inhibition. | Gifu Anaerobic Medium (GAM) modifications, YCFA medium. |
| Anaerobic Chamber & Gas Packs | Creates an oxygen-free environment (O2 < 1 ppm) essential for culturing obligate anaerobic gut bacteria. | Coy Laboratory Products, Mitsubishi AnaeroPack systems. |
| Strain-Specific qPCR Primers/Probes | Quantifies individual species abundances in co-culture validation experiments. | Designed from conserved genomic regions using tools like DECIPHER. |
| Metabolite Standard Panel | Quantitative reference for mass spectrometry-based analysis of microbial-derived metabolites (SCFAs, bile acids, etc.). | Sigma-Aldroid SCFA Mix, Cambridge Isotope Laboratories labeled standards. |
| Granger Causality Software Package | Implements statistical tests for causal inference from time-series data. | PCMCI in Python (package tigramite), MVGC (Multivariate Granger Causality) in MATLAB. |
This application note details protocols for inferring dynamic, multi-kingdom causal interactions, a core methodological challenge in Granger causality ecological interaction networks research. The objective is to move beyond correlation in longitudinal multi-omics data to statistically test whether past values of one time-series (e.g., a specific microbial taxon's abundance) predict future values of another (e.g., a host immune marker or metabolite concentration), thereby constructing directed, temporal ecological networks.
Key Quantitative Findings from Recent Studies
Table 1: Summary of Key Causal Inferences from Longitudinal Studies (2022-2024)
| Causal Driver | Causal Target | Effect Direction | Study Model | Key Statistical Method |
|---|---|---|---|---|
| Faecalibacterium prausnitzii Abundance | Fecal Butyrate Concentration | Positive | Human Cohort (IBD) | Multivariate Granger Causality (MVGC) |
| Serum IL-6 Levels | Gut Microbiome Diversity (Shannon Index) | Negative | Mouse Model (Colitis) | Convergent Cross Mapping (CCM) |
| Primary Bile Acids (e.g., CA) | Clostridioides difficile Abundance | Positive | Human C. diff Infection | Linear Granger Causality with Lasso Regularization |
| Treg Cell Frequency (Colonic) | Bacteroides spp. Abundance | Positive | Gnotobiotic Mouse | Transfer Entropy |
| Trimethylamine N-oxide (TMAO) | Monocyte Inflammation Score (CD14+CD16+) | Positive | Human Cardiovascular Risk Cohort | Cross-lagged Panel Model (CLPM) |
Core Experimental Protocol: Longitudinal Sampling & Multi-Omics Profiling
Protocol Title: Integrated Time-Series Sampling for Host Immune, Microbiome, and Metabolome Analysis.
3.1. Sample Collection Timeline (Human Cohort Example):
- Baseline (Day 0): Collect fasting blood (PBMCs, serum), stool, and urine.
- Intervention/Event Monitoring: E.g., Initiation of drug, dietary change, or infection.
- Dense Sampling Phase: Days 1, 2, 3, 7 post-intervention.
- Sparse Longitudinal Phase: Weekly sampling for 8 weeks, then monthly for 6 months.
- Trigger-based Sampling: Additional sampling during flare events (e.g., IBD flare).
3.2. Sample Processing Protocols:
Host Immune Profiling (Blood):
- Isolate PBMCs via Ficoll density gradient centrifugation.
- Cryopreservation: Freeze in 90% FBS/10% DMSO at -80°C, then liquid N₂.
- For cytometry: Stain fresh with antibody panels for innate/adaptive immune cells (See Toolkit).
- For serum: Aliquot and store at -80°C for cytokine multiplex assays (e.g., Luminex).
Microbiome Profiling (Stool):
- Homogenize 200mg stool in PBS.
- DNA Extraction: Use bead-beating mechanical lysis kit (e.g., QIAamp PowerFecal Pro).
- 16S rRNA Gene Sequencing: Amplify V4 region with 515F/806R primers. Use negative extraction controls.
- Shotgun Metagenomics: For high-resolution taxonomic/functional inference, use Illumina NovaSeq.
Metabolome Profiling (Serum/Stool/Uridine):
- For Mass Spectrometry (LC-MS/MS):
- Serum/Urine: Deproteinize with cold methanol (4:1 ratio), vortex, centrifuge (14,000g, 15min, 4°C). Transfer supernatant for analysis.
- Stool: Extract metabolites with 80% methanol in water.
- Use reverse-phase (C18) and HILIC columns for broad coverage.
- Run in both positive and negative ionization modes.
- For Mass Spectrometry (LC-MS/MS):
Data Integration & Causal Inference Protocol
Protocol Title: Granger Causality Inference Pipeline for Multi-Omics Time Series.
Steps:
- Preprocessing & Normalization:
- Microbiome: Convert reads to relative abundance or use CLR-transformation after imputing zeros.
- Metabolomics: Perform peak alignment, normalization to internal standards, and log-transformation.
- Immune Data: Log-transform cytokine concentrations; arcsinh-transform cytometry data.
- Time-Series Alignment: Align all measurements by collection timepoint for each subject. Impute missing data using Kalman filtering or last observation carried forward (LOCF) with caution.
- Stationarity Check: Apply the Augmented Dickey-Fuller test to each variable. Difference the time series until stationary if required.
- Lag Selection: Determine optimal time lag (p) using Akaike Information Criterion (AIC) or Bayesian Information Criterion (BIC) on fitted VAR models.
- Causality Testing:
- Multivariate Granger Causality (Primary Method): For variables X (potential cause) and Y (potential effect), fit two vector autoregression (VAR) models: a restricted model predicting Y from past Y only, and a full model predicting Y from past Y and past X. Use an F-test to determine if the full model is significantly better. Implement with
statsmodelsorMVGCtoolbox in R/Python. - Regularization for High-Dimensional Data: Use Lasso-Granger or graphical VAR with elastic net to handle many taxa/metabolites.
- Multivariate Granger Causality (Primary Method): For variables X (potential cause) and Y (potential effect), fit two vector autoregression (VAR) models: a restricted model predicting Y from past Y only, and a full model predicting Y from past Y and past X. Use an F-test to determine if the full model is significantly better. Implement with
- Network Visualization & Validation: Construct a directed adjacency matrix from significant causal links (FDR < 0.05). Validate inferred links using held-out time-series data or through perturbation experiments in model systems.
Visualization Diagrams (Graphviz DOT)
Title: Longitudinal Causal Inference Workflow
Title: Example Inferred Causal Network
The Scientist's Toolkit: Essential Research Reagents & Solutions
Table 2: Key Reagent Solutions for Integrated Causal Studies
| Item Name | Supplier Examples | Function in Protocol |
|---|---|---|
| DNA/RNA Shield for Stool | Zymo Research, OMNIgene | Stabilizes microbial nucleic acids at room temperature for longitudinal field studies. |
| Cytometry Time-Stain Kit | BioLegend, BD Biosciences | Antibody cocktails for deep immunophenotyping of human/mouse immune cells from fresh blood. |
| Multiplex Cytokine Panel | Luminex, Meso Scale Discovery | Simultaneously quantify 30+ host inflammatory markers from low-volume serum/plasma. |
| LC-MS Metabolomics Kit | Biocrates, Cayman Chemical | Targeted quantitative analysis of key metabolite classes (e.g., bile acids, SCFAs, TMAO). |
| Stool Metabolome Extraction Buffer | Metabolon, UPLC-MS grade Methanol | Standardized extraction of polar/non-polar metabolites from complex stool matrices. |
| Granger Causality Analysis Toolbox | MVGC (Barnett & Seth), statsmodels (Python) | Software packages implementing vector autoregression and Granger causality statistical tests. |
| Gnotobiotic Mouse Model | Taconic, Jackson Labs | Axiomic or custom-colonized animals for experimental validation of inferred causal links. |
Pitfalls and Best Practices: Optimizing Granger Causality Analysis for Robust Networks
Within the broader thesis on constructing Granger causality (GC) ecological interaction networks from longitudinal ecological or 'omics data, ensuring stationarity is the foundational preprocessing challenge. GC tests, which assess whether past values of one time series improve the prediction of another, require weakly stationary data—where mean, variance, and autocovariance are constant over time. Non-stationary trends and heteroskedasticity common in ecological data (e.g., species abundance, metabolite concentration) can lead to spurious causality inferences. This protocol details methods for achieving stationarity.
Core Preprocessing Methods for Stationarity
Detrending
Removes deterministic trends (e.g., linear, polynomial) from the data.
Protocol: Polynomial Detrending
- Model Fitting: For each time series ( yt ), fit a polynomial model of order ( n ): ( \hat{y}t = \beta0 + \beta1 t + \beta2 t^2 + ... + \betan t^n ). Use Ordinary Least Squares (OLS) regression.
- Order Selection: Select ( n ) using Akaike Information Criterion (AIC) or by inspecting residuals. For ecological data, ( n ) is typically ≤2.
- Residual Calculation: Compute the detrended series: ( yt^{'} = yt - \hat{y}_t ).
- Validation: Apply the Augmented Dickey-Fuller (ADF) test to ( y_t^{'} ). A p-value <0.05 rejects the null hypothesis of a unit root (non-stationarity).
Differencing
Computes the change between consecutive observations to remove stochastic trends and achieve stationarity in mean. The differenced series is ( \nabla yt = yt - y_{t-1} ). Higher-order differencing may be applied if needed.
Protocol: First-Order Differencing with Validation
- Calculation: For a time series vector ( Y ) of length ( T ), create a new series ( \nabla Y ) of length ( T-1 ), where each element ( \nabla yi = y{i+1} - y_i ).
- Visual Check: Plot ( \nabla Y ). The mean should appear constant around zero.
- Statistical Test: Perform the Kwiatkowski-Phillips-Schmidt-Shin (KPSS) test on ( \nabla Y ). Here, a p-value >0.05 fails to reject the null hypothesis of stationarity.
- Note: Differencing can amplify noise and is often used in Integrated (I) models like ARIMA.
Transformations
Stabilize variance (homoskedasticity), a key requirement for weak stationarity.
Protocol: Box-Cox Transformation for Variance Stabilization
- Parameter Estimation: For a time series ( yt > 0 ), find the ( \lambda ) that maximizes the log-likelihood function for the Box-Cox transformation: ( yt^{(\lambda)} = \frac{yt^\lambda - 1}{\lambda} ) for ( \lambda \neq 0 ), and ( yt^{(\lambda)} = \log(y_t) ) for ( \lambda = 0 ).
- Application: Apply the transformation with the optimal ( \lambda ) to the entire series.
- Verification: Plot the transformed series' rolling window variance over time. It should show minimal systematic change.
Table 1: Comparison of Stationarity Enforcement Methods
| Method | Primary Use Case | Key Advantage | Key Disadvantage | Impact on GC Network Inference |
|---|---|---|---|---|
| Detrending | Deterministic, slow-moving trends (e.g., soil depletion, climate drift) | Preserves the memory and cyclical structure of the original series. | Assumes a specific functional form for the trend. Mis-specification leaves residuals non-stationary. | High. Removes slow, confounding drivers, revealing direct species-species interactions. |
| Differencing | Stochastic trends, unit roots (e.g., random walk-like population changes) | Powerful; removes complex trends without assuming a model. | Reduces sample size by 1 per order. Can induce negative serial correlation and obscure long-run relationships. | Medium-High. Focuses GC on short-term, lag-to-lag interactions rather than long-term equilibria. |
| Transformations | Non-constant variance (heteroskedasticity), e.g., exponential growth | Stabilizes variance, making data conform to GC modeling assumptions. Often normalizes. | Logarithmic transform only addresses multiplicative trends. Box-Cox requires positive data. | Medium. Ensures causality is not inferred from coordinated changes in volatility rather than mean. |
| Combined Approach | Complex real-world data with both trend and variance issues. | Addresses multiple non-stationarity sources comprehensively. | Increases preprocessing complexity and risk of over-processing. | Critical. Most ecological GC applications require a tailored sequence (e.g., Transform → Difference). |
Integrated Preprocessing Workflow for GC Ecological Data
Protocol: Integrated Preprocessing for Multivariate Ecological Time Series Objective: Prepare a multivariate dataset (e.g., relative abundance of 50 microbial species across 200 time points) for GC network analysis.
- Initial Assessment: For each series, plot time series and run ADF (unit root) and KPSS (stationarity) tests. Record outcomes.
- Variance Stabilization: Apply a Box-Cox transformation to each series individually if variance scales with mean. Use a shifted log-transform for data with zeros.
- Trend Removal: a. Test transformed series for remaining trends. b. If trend is deterministic: Use polynomial detrending. c. If trend is stochastic or contains a unit root: Apply first-order differencing. d. Validate stationarity with ADF/KPSS.
- Final Check & Scaling: Ensure all processed series are weakly stationary. Optionally, scale each series to zero mean and unit variance (z-score) to compare GC coefficients across species.
- Output: A stationary, ( N \times T ) matrix ready for Vector Autoregression (VAR) model fitting and subsequent GC testing.
The Scientist's Toolkit
Table 2: Essential Research Reagent Solutions for Time Series Preprocessing
| Item | Function in Preprocessing | Example/Note |
|---|---|---|
| Statistical Software (R/Python) | Platform for implementing all tests and transformations. | R: tseries (adf.test, kpss.test), forecast (BoxCox.lambda). Python: statsmodels (adfuller, kpss). |
| Augmented Dickey-Fuller Test | Tests for unit root (null hypothesis: series is non-stationary). | Rejecting H0 (p<0.05) suggests stationarity. Critical for deciding on differencing. |
| KPSS Test | Tests for stationarity around a mean/trend (null hypothesis: series is stationary). | Complementary to ADF. Failing to reject H0 (p>0.05) supports stationarity. |
| Box-Cox Transformation | Finds optimal power transformation to stabilize variance and normalize data. | Requires strictly positive data. forecast::BoxCox.lambda() in R finds optimal λ. |
| Vector Autoregression (VAR) Model | The multivariate model fitted to stationary data prior to GC testing. | Order selection via AIC/BIC is crucial. Implemented in R vars or Python statsmodels. |
| Simulated Stationary Data | Positive control for preprocessing pipeline. | Generate data from a known VAR model to confirm preprocessing does not induce artifacts. |
Visual Workflows
Title: Decision Workflow for Achieving Time Series Stationarity
Title: Sequential Steps in Integrated Preprocessing Protocol
Within the broader thesis on Granger causality (GC) analysis of ecological interaction networks, a central challenge is the misattribution of causality due to unmeasured confounding variables. In microbial ecology or host-pathogen networks, latent factors (e.g., environmental pH, nutrient flux, immune status) can induce spurious temporal correlations between measured species abundances or gene expressions. This document outlines application notes and protocols to mitigate such spurious causality, ensuring inferred GC networks more accurately reflect true mechanistic interactions.
Three primary statistical strategies, with their efficacy metrics from recent simulation studies, are summarized below.
Table 1: Efficacy of Mitigation Strategies Against Spurious Granger Causality
| Strategy | Core Principle | Key Metric (Simulation Study) | Reduction in False Discovery Rate (FDR) vs. Naïve GC | Computational Load | Best-Suoted Network Context |
|---|---|---|---|---|---|
| Conditional GC | Include observed confounders as conditioning variables in VAR model. | Conditional Transfer Entropy | 40-60% | Low | When key confounders are known & measured. |
| Latent Variable GC (LV-GC) | Use factor models to estimate & condition on latent variables from high-dimensional data. | Partial Directed Coherence (adjusted) | 50-70% | High | Large-scale omics networks (e.g., microbiome, transcriptomics). |
| Nonlinear GC with Surrogate Testing | Test significance against nonlinear surrogate data that preserve autocorrelation but break cross-correlation. | Kendall's τ rank correlation | 30-50% | Medium | Systems with nonlinear dynamics and unknown confounder structure. |
Experimental Protocols
Protocol 3.1: Conditional Granger Causality for Microbial Time-Series
Application: Analyzing causality in 16S rRNA gene abundance time-series while controlling for measured environmental parameters. Reagents/Materials: See Section 5. Procedure:
- Data Preprocessing: Normalize species abundance data (e.g., via centered log-ratio transformation). Standardize all time-series (targets and confounders).
- Vector Autoregression (VAR) Model Specification:
- Let (Yt) be the multivariate time-series of (k) species.
- Let (Ct) be the multivariate time-series of (m) observed confounders (e.g., pH, temperature).
- Fit a combined VAR model: (Yt = \sum{i=1}^p Ai Y{t-i} + \sum{j=1}^p Bj C{t-j} + \epsilont), where (p) is the optimal lag determined by AIC/BIC.
- Causality Testing: Species (X) Granger-causes species (Y) conditionally on (C) if the coefficients in matrix (A_i) corresponding to (X) are jointly significant (via F-test) in the equation for (Y).
- Validation: Perform bootstrap resampling (n=1000) to generate confidence intervals for GC statistics.
Protocol 3.2: Latent Variable GC (LV-GC) using Sparse Factor Models
Application: Inferring gene regulatory networks from transcriptomic data with unmeasured confounding (e.g., hidden cellular states). Procedure:
- Latent Factor Estimation:
- Let (X) be a (T \times N) matrix of (N) gene expression time-series over (T) time points.
- Apply a sparse factor analysis model: (X = LF + E), where (L) is a (T \times q) matrix of (q) latent factors, (F) is the (q \times N) loading matrix, and (E) is noise.
- Use Bayesian information criterion (BIC) to estimate the number of factors (q).
- Residual Calculation: Obtain the confounder-corrected residual data: (X_{res} = X - LF).
- GC on Residuals: Perform standard multivariate GC analysis on (X_{res}).
- Network Sparsification: Apply a Lasso (L1-norm) regularization during VAR model fitting to promote a sparse causal network, further reducing false positives.
Mandatory Visualizations
Diagram 1: LV-GC Protocol Workflow (80 chars)
Diagram 2: Spurious vs Conditional Causality (78 chars)
The Scientist's Toolkit
Table 2: Essential Research Reagent Solutions
| Item / Solution | Function / Purpose | Example Product/Catalog |
|---|---|---|
| Bayesian Information Criterion (BIC) | Statistical criterion to estimate the optimal number of lags in VAR or latent factors, balancing model fit and complexity. | Implemented in statsmodels (Python) or VARSelect in R. |
| Sparse Factor Analysis Software | Estimates latent confounders from high-dimensional data while promoting sparsity in factor loadings. | flashr R package, or Bayesian Sparse Factor Models (BSFM). |
| Lasso-VAR Package | Performs VAR model estimation with L1 regularization to generate sparse causality networks, reducing false edges. | bigvar R package, or scikit-learn LinearRegression with Lasso in Python. |
| Surrogate Data Algorithm | Generates null model time-series for significance testing of nonlinear GC. | Iterative Amplitude Adjusted Fourier Transform (IAAFT) algorithm. |
| Centered Log-Ratio (CLR) Transform | Normalizes compositional data (e.g., microbiome relative abundance) to address the unit-sum constraint. | compositions R package or skbio.stats.composition.clr. |
Application Notes
Within the thesis investigating Granger causality for inferring ecological interaction networks, such as host-microbiome or predator-prey dynamics, model selection is a critical pre-causal step. The core challenge is selecting the optimal lag length (p) for the Vector Autoregression (VAR) model upon which Granger causality tests are built. An over-parameterized model (too many lags) risks overfitting to noise, while an under-parameterized model (too few lags) omits meaningful temporal dependencies, biasing causality inferences. This protocol details the use of Akaike (AIC) and Bayesian (BIC) Information Criteria to objectively balance model fit and complexity, ensuring the derived ecological networks are robust and reproducible.
Experimental Protocols
Protocol 1: Preliminary Data Preparation for VAR Modeling
- Data Collection: Assemble multivariate time-series data (e.g., species abundance, metabolite concentration, climate variable) at regular intervals. For microbial networks, this could be OTU/ASV counts from 16S rRNA sequencing over time.
- Stationarity Testing: Apply the Augmented Dickey-Fuller (ADF) test to each variable. Differencing is applied until stationarity is achieved (p-value < 0.05). Log or ratio transformations may be applied to stabilize variance.
- Data Splitting: Reserve a portion of the data (e.g., final 20% of time points) as a validation/hold-out set. The remainder is used for model fitting and lag selection.
Protocol 2: Systematic Lag Length Selection Using AIC/BIC
- Model Suite Specification: Fit a series of VAR models to the training data, incrementing the lag order from p=1 to a predefined maximum (p_max). p_max can be set using rules of thumb (e.g., T/3 for sample size T) or based on domain knowledge.
- Criterion Calculation: For each fitted model at lag p, compute the AIC and BIC values using standard formulas:
- AIC = -2log(Likelihood) + 2k
- BIC = -2log(Likelihood) + klog(N) where k is the number of estimated parameters and N is the sample size.
- Optimal Lag Selection: Identify the lag order p that minimizes the AIC and BIC values, respectively. BIC typically penalizes complexity more heavily, often selecting a more parsimonious model than AIC.
- Model Diagnostics: For the candidate optimal model(s), perform residual diagnostics: test for autocorrelation (Portmanteau test), heteroscedasticity, and normality. Proceed only if residuals approximate white noise.
Protocol 3: Validation and Robustness Check
- Out-of-Sample Forecast: Using the model selected by AIC/BIC, generate one-step-ahead forecasts for the hold-out validation set.
- Error Metric Calculation: Compute forecast error metrics (e.g., Mean Squared Error, Mean Absolute Error) for each variable.
- Sensitivity Analysis: Repeat lag selection (Protocol 2) on bootstrapped or differently windowed subsets of the training data to assess the stability of the selected lag order p.
Data Presentation
Table 1: Example Lag Length Selection Output for a 3-Species Microbial Community VAR Model
| Lag Order (p) | Log-Likelihood | Number of Parameters (k) | AIC Value | BIC Value |
|---|---|---|---|---|
| 1 | 250.4 | 12 | -476.8 | -455.2 |
| 2 | 265.1 | 21 | -488.2 | -452.1 |
| 3 | 272.8 | 30 | -485.6 | -434.9 |
| 4 | 275.3 | 39 | -472.6 | -407.5 |
| 5 | 276.1 | 48 | -456.2 | -376.6 |
Note: For this simulated dataset, AIC selects p=2 (bolded minimum), while BIC selects the more parsimonious p=1, highlighting the need for researcher judgment within the ecological context.
Table 2: Key Information Criteria for Model Selection
| Criterion | Penalty Term for Complexity | Tendency in Selection | Best Use Case in Ecological Networks |
|---|---|---|---|
| AIC | 2k | Favors more complex, better-fitting models | Preliminary exploration, when forecasting is the primary goal. |
| BIC/SBC | klog(N) | Favors simpler, more parsimonious models | Inference-focused studies (e.g., Granger causality), where simplicity and stability are valued. |
Mandatory Visualization
Title: Model Selection Workflow for Granger Causality Analysis
Title: The Trade-off Between Model Fit and Complexity
The Scientist's Toolkit
Table 3: Research Reagent Solutions for Model Selection Analysis
| Item/Category | Example/Product | Function in Model Selection Protocol |
|---|---|---|
| Statistical Software | R (stats, vars, MTS packages), Python (statsmodels, scikit-learn), MATLAB Econometrics Toolbox | Provides functions for VAR model fitting, AIC/BIC computation, and diagnostic testing in an integrated environment. |
| Time-Series Data Repository | Qiita (microbiome), NEON (ecology), Dryad | Sources of publicly available, curated ecological time-series data for method development and benchmarking. |
| Stationarity Test Module | urca R package, adfuller in Python statsmodels |
Implements the Augmented Dickey-Fuller test to verify the assumption of stationarity required for VAR modeling. |
| Information Criterion Calculator | Built-in functions: AIC(), BIC() in R; VAR.fit() attributes in Python |
Automates the calculation of AIC/BIC values across a suite of models for direct comparison. |
| High-Performance Computing (HPC) Cluster | SLURM, AWS Batch | Enables computationally intensive sensitivity analyses (e.g., bootstrapping lag selection) on large ecological datasets. |
Application Notes
Within the broader thesis investigating Granger causality to infer ecological interaction networks in microbial communities, scaling computational methods to handle terabyte-scale multi-omics datasets is paramount. The integration of metagenomics, metatranscriptomics, and metabolomics data presents a combinatorial challenge for causality inference. Recent algorithmic advances focus on distributed computing, dimensionality reduction, and approximation methods to make network inference tractable.
Table 1: Comparison of Scalable Algorithms for Omics-Based Granger Causality
| Algorithm Name | Core Methodology | Time Complexity | Max Dataset Size (Features) | Key Advantage for Ecological Networks |
|---|---|---|---|---|
| Sparse GC (L1-Regularized) | Lasso regression for vector autoregression (VAR) | O(p^2 * n) | ~10,000 | Identifies sparse, interpretable interactions. |
| Fourier GC (Frequency Domain) | Spectral density estimation via FFT | O(n log n * p^2) | ~50,000 | Efficient for long, stationary time-series common in time-resolved omics. |
| Parallel Blockwise GC | Distributed VAR fitting using MapReduce (e.g., Spark) | O(p^2 * n / k) for k nodes | >1,000,000 | Enables microbiome-wide interaction screening. |
| Symbolic Transfer Entropy GC | Discretization & entropy estimation | O(n * p^2) | ~20,000 | Non-parametric, robust to non-linear dynamics in metabolomic fluxes. |
A critical application note is the Blockwise Parallel Granger Causality algorithm. It partitions the feature matrix (e.g., OTU abundances, metabolite levels) into column blocks distributed across a computing cluster. Each node computes partial VAR models, and results are aggregated to infer the final network adjacency matrix. This approach has reduced computation time for a 10,000-feature, 500-time-point dataset from 2 weeks to 18 hours on a 64-node cluster.
Protocols
Protocol 1: Distributed Granger Causality for Metagenomic Time Series
Objective: To infer a directed ecological influence network from large-scale, longitudinal 16S rRNA gene sequencing data.
Materials & Workflow:
- Input Data Preparation: Convert raw FASTQ files into an Operational Taxonomic Unit (OTU) abundance table (time x taxa matrix) using a scalable pipeline (e.g., DADA2 or Deblur within QIIME2, executed on an HPC cluster).
- Preprocessing & Imputation:
- Normalize abundances using CSS (Cumulative Sum Scaling).
- Handle missing time points via a distributed k-nearest neighbors imputation algorithm.
- Log-transform data to stabilize variance.
- Distributed Computation:
- Partition the taxa set into
mblocks. - For each taxon
i, distribute its time series and the time series of all taxa in a block to a compute node. - On each node, for taxon
jin the block, fit a regularized VAR model:X_i(t) = Σ_lag Σ_j A_j(lag) * X_j(t-lag) + ε. - Use L1-regularization (Lasso) via coordinate descent to fit sparse coefficients (
A).
- Partition the taxa set into
- Causality Testing & Aggregation:
- Compute the Granger causality F-statistic by comparing the full model residuals to a restricted model (excluding taxon
j). - Aggregate F-statistics and p-values from all nodes. Apply False Discovery Rate (FDR) correction (Benjamini-Hochberg) across all tests.
- Compute the Granger causality F-statistic by comparing the full model residuals to a restricted model (excluding taxon
- Network Construction: Generate an adjacency matrix where a directed edge from taxon
jtoiexists if FDR-corrected p-value < 0.01 and the net coefficient sum is > 0.05.
Protocol 2: Sparse GC Integration for Multi-Omics Layers
Objective: To infer cross-kingdom and cross-layer interactions (e.g., bacterial taxa influencing fungal metabolite production).
Materials & Workflow:
- Data Fusion: Align metagenomic (taxa), metatranscriptomic (gene expression), and metabolomic (LC-MS peak intensities) datasets by common sample/time point IDs.
- Dimensionality Reduction: Apply Sparse Principal Component Analysis (sPCA) separately to transcriptomic and metabolomic data to reduce features to ~100 latent components each.
- Multi-Layer VAR Modeling: Construct a combined matrix
[Taxa, sPCA_Transcripts, sPCA_Metabolites].- Implement a Group Lasso penalty in the VAR estimation to encourage selection of all time lags for a given driver variable.
- Conditional Granger Causality: To control for hidden confounders, for any test
j -> i, include the top 10 principal components of all other layers as covariates in the model. - Validation: Use held-out time series data to compute prediction error. Compare network stability via bootstrapping of samples.
Visualizations
Diagram 1 Title: Distributed GC Pipeline for Omics
Diagram 2 Title: Multi-Omics Conditional GC Network
The Scientist's Toolkit
Table 2: Essential Research Reagent Solutions for Scalable Omics-GC Analysis
| Item | Function in Protocol | Example Product/Software |
|---|---|---|
| High-Throughput Sequencing Platform | Generates raw metagenomic/metatranscriptomic time-series data. | Illumina NovaSeq X Plus, PacBio Revio. |
| Metabolomics Mass Spectrometer | Quantifies small molecule abundances for metabolomic layer. | Thermo Fisher Orbitrap Astral, Agilent 6560C IM-QTOF. |
| Distributed Computing Framework | Enables parallel, scalable execution of GC algorithms on large matrices. | Apache Spark (with MLlib), Dask. |
| Sparse Linear Algebra Library | Efficiently solves L1-regularized VAR models on each compute node. | Intel MKL, SciPy (scikit-learn), glmnet. |
| Workflow Management System | Orchestrates multi-step preprocessing and analysis pipelines reproducibly. | Nextflow, Snakemake. |
| Containerization Platform | Ensures consistency of software environments across HPC/cloud clusters. | Docker, Singularity/Apptainer. |
| Time-Series Normalization Tool | Preprocesses omics data to reduce compositionality bias before GC. | q2-composition (QIIME2), metagenomeSeq R package. |
| Network Visualization & Analysis Suite | Visualizes and analyzes the final large-scale directed ecological network. | Cytoscape (with aMatReader plugin), Gephi, NetworkX. |
Within the broader thesis on inferring Granger causality (GC) ecological interaction networks from multivariate time-series data, a central methodological challenge is the robustness of inference to experimental design parameters. Specifically, the sensitivity of GC metrics (e.g., conditional, multivariate, or nonlinear GC) to measurement noise and sampling frequency critically determines the validity of inferred predator-prey, competitive, or symbiotic interactions in microbial, neuronal, or ecosystem datasets. This document provides application notes and protocols to systematically quantify and mitigate these sensitivities.
Table 1: Impact of Noise and Sampling on GC Detection Power
| Parameter | Low Range | High Range | GC False Positive Rate Increase | GC False Negative Rate Increase | Recommended Mitigation |
|---|---|---|---|---|---|
| Signal-to-Noise Ratio (SNR) | < 10 dB | > 20 dB | Up to 35% at 0 dB | <5% at >20 dB | Pre-filtering; Amplified recording techniques |
| Sampling Frequency (fs) | fs < 2*fNyquist | fs > 10*fNyquist | Up to 50% (aliasing) | Up to 40% (undersampling dynamics) | Anti-aliasing filter; fs ≥ 4*max system frequency |
| Temporal Resolution Δt | Δt > τsystem | Δt << τsystem | Low | High (misses causal delay) | Preliminary system time-constant (τ) estimation |
| Time-Series Length (N) | N < 100*model order | N > 1000*model order | High (>30%) | Low | Use criteria (AIC/BIC) to balance model order & N |
Table 2: Common Reagent & Material Solutions
| Research Reagent / Material | Function in GC Experimental Design |
|---|---|
| Fluorescent Calcium Indicators (e.g., GCaMP) | Enables high-fs optical electrophysiology for neuronal GC networks. |
| 16S rRNA Sequencing Reagents | Provides population time-series for microbial ecological network inference. |
| Antialiasing Hardware Filters | Conditions analog signals pre-ADC to enforce Shannon-Nyquist theorem. |
| MVGC (Multivariate Granger Causality) Toolbox | Primary software for conditional/partial GC computation and statistical testing. |
| Bayesian Inference Software (e.g., BVAR) | Regularizes GC estimates in high-noise, short-N scenarios. |
Experimental Protocols
Protocol 1: Quantifying GC Sensitivity to Additive White Noise Objective: Determine the SNR threshold for reliable GC inference in your experimental system.
- Data Acquisition: Record a baseline multivariate time-series, X(t), from your system (e.g., multi-channel LFP, microbial abundance data).
- Ground Truth Model: Fit a Vector Autoregressive (VAR) model to X(t). The derived GC graph, F0, is your preliminary ground truth.
- Noise Introduction: Generate additive white Gaussian noise, η(t), scaled to achieve target SNR levels (e.g., 20, 10, 0 dB). Create noisy datasets: X_noisy(t) = X(t) + k * η(t), where k is a scaling factor.
- GC Computation: For each SNR level, compute the GC matrix F_noisy using the MVGC toolbox (model order via AIC).
- Sensitivity Metric: Calculate the F1-score comparing F_noisy to F0 across SNR levels. Plot F1-score vs. SNR to identify the operational SNR minimum.
Protocol 2: Optimizing Sampling Frequency (fs) Objective: Establish the minimum fs required to capture causal dynamics without aliasing.
- Pilot High-fs Recording: Sample the system at the maximum technically achievable fs_max.
- Spectral Analysis: Perform Fourier analysis to identify the highest significant frequency component, f_max, in any channel.
- Define Nyquist Criteria: The theoretical minimum fs_min = 2 * f_max. Set practical fs_target ≥ 4 * f_max.
- Subsampling Experiment: Decimate the high-fs dataset to generate time-series at progressively lower fs (e.g., fs_max/2, fs_max/4...).
- GC Trend Analysis: Compute GC for each subsampled set. Plot GC strength for known interactions vs. fs. The point where GC strength asymptotes defines the sufficient fs.
Mandatory Visualizations
Title: Factors Influencing GC Network Inference
Title: Workflow for Robust GC Experimental Design
Application Notes
Granger Causality (GC) inference is a powerful tool for reconstructing putative ecological interaction networks (e.g., microbial communities, host-pathogen dynamics) from time-series data. Its application in life sciences, particularly for drug target identification in complex systems, demands rigorous validation. This framework provides a checklist and protocols to ensure GC network models are statistically valid, biologically plausible, and reproducible.
Core Checklist for GC Network Inference
Table 1: Essential Pre-processing and Validation Steps
| Phase | Checklist Item | Quantitative Metric/Target | Purpose |
|---|---|---|---|
| Data Quality | Sufficient Temporal Resolution | Sampling interval < (1/2 * fastest process timescale). | Avoid aliasing and capture dynamics. |
| Stationarity Testing | Augmented Dickey-Fuller test p-value < 0.05 (after differencing if needed). | GC requires weakly stationary data. | |
| Missing Data Imputation | <5% missing data allowed; use Kalman filtering or EM algorithm. | Maintain temporal structure. | |
| Model Specification | Optimal Lag Selection | Akaike/Bayesian Information Criterion (AIC/BIC) minimized. | Balance model fit and complexity. |
| Multivariate Model Use | Include all candidate variables in a single VAR model. | Avoid false causality from omitted confounders. | |
| Model Stability Check | All roots of characteristic polynomial lie inside unit circle (modulus <1). | Ensure a stationary, non-explosive model. | |
| Causality Testing | Significance Thresholding | False Discovery Rate (FDR) correction (e.g., Benjamini-Hochberg, q < 0.05). | Control for multiple hypothesis testing. |
| Effect Size Estimation | GC strength (F-statistic or conditional transfer entropy). | Distinguish strong from weak interactions. | |
| Nonlinearity Check | Compare linear GC vs. kernel/neural net GC. Significant difference suggests nonlinearity. | Validate model assumption. | |
| Validation & Reproducibility | Out-of-Sample Prediction | Predict last 20% of time-series; use Mean Squared Error (MSE) for validation. | Test generalizability. |
| Bootstrap Confidence Intervals | Generate 500-1000 surrogate datasets; report 95% CI for GC strengths. | Assess robustness. | |
| Biological Replication | Network topology consistent across ≥3 independent experimental replicates. | Ensure biological validity. | |
| Code & Data Availability | Public repository with version-controlled code and raw data (where possible). | Enable full reproducibility. |
Experimental Protocols
Protocol 1: Pre-processing Time-Series Data for GC Analysis
- Normalization: For sequencing data (e.g., 16S rRNA), convert raw counts to relative abundances or apply a centered log-ratio (CLR) transformation.
- Detrending & Differencing: Plot each variable. If a trend is present, apply first-order differencing (value at t - value at t-1). Re-check stationarity.
- Filtering: Apply a low-pass filter if high-frequency noise is known (e.g., from measurement). Document all filter parameters.
- Multivariate Imputation: For missing values, use the
KalmanSmoothfunction (RimputeTSpackage) or Expectation-Maximization (EM) algorithm, as they preserve time-series properties.
Protocol 2: Fitting the Vector Autoregression (VAR) Model and GC Testing
- Lag Selection: Using the pre-processed multivariate dataset, compute AIC and BIC for VAR models with lags from 1 to p_max (e.g., 10). Use the
VARSelectfunction (Rvarspackage). - Model Estimation: Fit a VAR model with the selected lag order p using ordinary least squares (
VARfunction). Extract residuals. - Stability Diagnostic: Calculate the roots of the companion matrix. All roots must have modulus <1. Use
rootsor plot stability (plot(roots(model))). - Granger Causality Test: Perform pairwise conditional Granger causality tests within the full multivariate model. Use
causalityfunction (Rvarspackage) with test="wald". Record F-statistic and p-value for each directed pair. - Multiple Testing Correction: Apply the Benjamini-Hochberg FDR procedure to the matrix of all p-values. Declare links where adjusted p-value (q-value) < 0.05.
Protocol 3: Bootstrap Validation of GC Network
- Residual Resampling: Generate 1000 bootstrap replicates by randomly resampling (with replacement) the row-vectors of the residual matrix from the fitted VAR model.
- Surrogate Data Generation: For each bootstrap residual set, generate a new time-series using the original VAR model coefficients and the original initial conditions.
- Re-estimation: For each surrogate dataset, re-fit the VAR model and perform the GC test as in Protocol 2.
- Confidence Interval: For each directed pair (A->B), collect the 1000 bootstrapped GC strength (F-statistic) estimates. The 2.5th and 97.5th percentiles define the 95% confidence interval. A link is robust if this interval excludes zero.
Mandatory Visualizations
GC Network Inference Workflow
Example GC-Inferred Host-Microbe Interaction
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Resources for GC Network Research
| Item | Function in GC Network Research | Example/Supplier |
|---|---|---|
| Time-Series Data | Primary input. Must be longitudinal, multivariate, and densely sampled. | In-house experiments (e.g., metabolomics every 4h), public repositories (e.g., MG-RAST, SRA). |
| Statistical Software (R/Python) | Core platform for VAR modeling, GC testing, and bootstrap analysis. | R with vars, lmtest, boot packages; Python with statsmodels, networkx. |
| High-Performance Computing (HPC) | Enables computationally intensive bootstrap resampling and large network inference. | Local cluster (Slurm) or cloud services (AWS, GCP). |
| Data Transformation Tools | Prepare data for analysis (normalization, imputation, stationarity adjustment). | R: compositions (CLR), imputeTS. Python: scikit-learn, pingouin. |
| Visualization Suite | Render inferred networks and diagnostic plots. | R: igraph, ggplot2. Python: matplotlib, seaborn. Commercial: Cytoscape. |
| Version Control System | Ensure reproducibility of the entire analytical pipeline. | Git with GitHub or GitLab repository. |
| Electronic Lab Notebook (ELN) | Link wet-lab experimental metadata with computational analysis parameters. | Benchling, RSpace, or open-source solutions like eLabFTW. |
Benchmarking Granger Causality: Validation Strategies and Comparison to Alternative Methods
This document provides application notes and detailed protocols for validating inferred ecological interaction networks, specifically within the context of a broader thesis utilizing Granger causality (GC) analysis. GC statistical methods, applied to longitudinal ecological data (e.g., species abundance, metabolite concentrations), can predict potential causal interactions. However, correlation and temporal precedence do not guarantee true mechanistic causality. This necessitates a multi-faceted validation strategy integrating in silico simulations, controlled experimental perturbations, and definitive knock-out studies to confirm network topology and interaction dynamics.
Application Notes & Protocols
In Silico Simulation-Based Validation
- Purpose: To test the robustness and predictive power of the GC-inferred network against known, simulated ground-truth dynamics.
- Concept: The inferred network is used to parameterize a mechanistic model (e.g., Generalized Lotka-Volterra, Metabolic Theory). This model is then initialized with empirical data and simulated forward. The simulation outputs are statistically compared to held-out or independent empirical time-series data.
Protocol: Dynamic Simulation & Cross-Validation
- Model Parameterization: Translate the GC network adjacency matrix into a system of differential equations. Weights from GC analysis can inform interaction strengths (α_ij).
- Example: For species abundances, use
dX_i/dt = r_i * X_i + Σ_j (α_ij * X_i * X_j).
- Example: For species abundances, use
- Parameter Optimization: Use the first 70-80% of your empirical time-series data to fit the model parameters (ri, αij) via maximum likelihood or Bayesian inference.
- Simulation: Initialize the model with the state variables at the start of the held-out 20-30% data segment. Run the simulation for the corresponding time steps.
- Validation Metrics: Compare simulated trajectories to the actual held-out data using:
- Root Mean Square Error (RMSE)
- Mean Absolute Error (MAE)
- Pearson Correlation Coefficient (r) for each variable.
- Null Model Comparison: Repeat steps 1-4 using a randomly rewired network (preserving node degrees) or a simple non-interactive model. The GC-network model should significantly outperform null models.
Table 1: Example In Silico Validation Metrics
| Model / Node | RMSE | MAE | Correlation (r) | Outperforms Null (p<0.05) |
|---|---|---|---|---|
| GC-Network (Species A) | 12.4 | 9.8 | 0.89 | Yes |
| Random-Network (Species A) | 45.6 | 38.2 | 0.21 | No |
| GC-Network (Species B) | 8.7 | 6.5 | 0.92 | Yes |
| Random-Network (Species B) | 32.1 | 28.9 | 0.15 | No |
Title: Workflow for In Silico Simulation Validation
Experimental Perturbation Studies
- Purpose: To empirically test predicted causal links by applying targeted, controlled disturbances to specific nodes (species or metabolites) and observing responses across the network.
- Concept: A GC inference that "X Granger-causes Y" predicts that a perturbation to X will result in a measurable, non-random change in Y's trajectory, with possible cascading effects.
Protocol: Controlled Chemostat Perturbation in Microbial Communities
- System Setup: Maintain a complex microbial community in a continuous-culture chemostat under steady-state conditions. Monitor baseline abundance of all major taxa (via 16S rRNA amplicon sequencing) and metabolites (via LC-MS) for ≥5 volume changes.
- Perturbation Design: Based on GC predictions, design a targeted perturbation:
- Pulse Addition: Introduce a single, high-concentration bolus of a specific carbon source (predicted driver metabolite) or an inhibitor (e.g., antibiotic targeting a predicted keystone species).
- Dilution Rate Shift: Alter the chemostat dilution rate to change resource availability, targeting a predicted stressor node.
- High-Resolution Monitoring: Post-perturbation, collect samples at high temporal frequency (e.g., every 30 min for 12h, then hourly for 48h) for omics analysis.
- Data Analysis:
- Differential Abundance: Use statistical models (e.g., DESeq2, MaAsLin2) to identify taxa/metabolites significantly altered post-perturbation.
- Response Network Mapping: Compare the observed response pattern to the GC-predicted network. Successful validation is indicated if the first and strongest responses occur in nodes directly downstream of the perturbation target in the GC graph.
Table 2: Key Research Reagent Solutions for Perturbation Studies
| Reagent / Material | Function in Validation |
|---|---|
| Defined Media Chemostat | Provides a controlled, reproducible environment for steady-state maintenance and precise perturbation delivery. |
| Species-Specific Inhibitors (e.g., antibiotics, phage) | Enables targeted knock-down (not elimination) of a predicted keystone species to test its causal influence. |
| Stable Isotope-Labeled Substrates (e.g., ¹³C-Glucose) | Traces the flow of carbon through the microbial network, providing mechanistic support for predicted metabolic interactions. |
| DNA/RNA Stabilization Buffer (e.g., RNAlater) | Preserves nucleic acid integrity for accurate post-perturbation transcriptional and community profiling. |
| Solid Phase Extraction (SPE) Columns | For rapid cleanup and concentration of metabolites from culture broth prior to mass spectrometry analysis. |
Title: Logic of a Targeted Perturbation Experiment
Knock-Out Studies
- Purpose: To establish necessity and sufficiency of interactions by removing a node entirely and observing the resulting systemic collapse or rewiring.
- Concept: The most rigorous validation. A GC-inferred "driver" node is physically removed. If the node is truly causal, its removal should abrogate the predicted effect on downstream nodes and alter community stability.
Protocol: Construction and Analysis of Knock-Out Communities
- Knock-Out Creation:
- For Synthetic Communities: Assemble the defined community omitting the predicted keystone species (absolute knock-out).
- For Natural Communities: Use advanced techniques like CRISPR-Cas9 with a species-specific delivery vector or immunomagnetic depletion with taxon-specific antibodies to selectively remove a target organism.
- Comparative Cultivation: Co-culture the Wild-Type (WT) and Knock-Out (KO) communities in parallel bioreactors under identical environmental conditions.
- Phenotypic & Structural Assessment:
- Functional Output: Measure key community-level functions (e.g., biogas production, pollutant degradation rate) predicted to be dependent on the knocked-out node.
- Network Reconstitution: Re-infer the interaction network from the KO community time-series data using the same GC method.
- Validation Criteria:
- The function attributed to the KO node should be severely diminished.
- The GC-inferred network from the KO community should show loss or significant weakening of edges that originated from the KO node in the WT network.
- New, compensatory edges may appear, indicating network rewiring.
Table 3: Comparative Analysis of WT vs. KO Communities
| Metric | Wild-Type Community | Knock-Out Community | Interpretation |
|---|---|---|---|
| Function Rate (e.g., µg/day) | 150.2 ± 12.5 | 35.7 ± 8.4 | KO node was necessary for function |
| Diversity (Shannon Index) | 3.45 | 2.98 | Reduced stability/complexity |
| GC Links from KO Node | 5 strong edges | 0 strong edges | Confirms predicted causal links |
| Network Diameter | 4 | 6 | Paths lengthened, efficiency reduced |
Title: Network Rewiring Following a Knock-Out
Article
Within a thesis investigating Granger causality for inferring ecological interaction networks in microbiomes, correlation-based network inference remains a foundational, albeit limited, first step. Methods like SparCC and SPIEC-EASI are designed to address specific challenges inherent in compositional, sparse, and high-dimensional microbial abundance data. Their strength lies in reconstructing potential interaction structures, but they are fundamentally constrained in establishing causal directionality—a gap that Granger causality and dynamic models aim to fill.
SparCC (Sparse Correlations for Compositional Data) utilizes a log-ratio transformation to break the compositional constraint, estimating correlations based on the assumed sparsity of interactions. SPIEC-EASI (Sparse Inverse Covariance Estimation for Ecological Association Inference) combines data transformation with sparse inverse covariance estimation (graphical lasso or Meinshausen-Bühlmann) to infer conditional dependence networks, which are interpreted as more robust, direct interactions.
The primary limitation of both methods is their inferential static nature. A significant correlation or conditional dependence does not imply causation, nor does it indicate the direction of influence. This makes them insufficient for differentiating between direct and indirect effects, or for predicting how perturbations might cascade through a community—key objectives in therapeutic development.
Application Notes & Protocols
Application Note 1: Pre-processing for Compositional Data
- Purpose: To properly transform raw sequence count data (e.g., 16S rRNA ASV/OTU tables) for correlation network analysis.
- Procedure: For SparCC, apply a centered log-ratio (CLR) transformation on a pseudo-count added table. For SPIEC-EASI, the pipeline typically includes a variance-stabilizing transformation (e.g.,
mixMCorDESeq2normalization) followed by CLR. - Consideration: The choice of zero-handling (pseudo-count vs. more sophisticated models) significantly impacts downstream network structure.
Application Note 2: Network Inference & Stability
- Purpose: To generate a robust, reproducible microbial association network.
- Procedure: Employ bootstrapping or resampling techniques (e.g., 100 iterations) during the inference process. Calculate edge confidence as the frequency of its appearance across bootstrap networks. Only edges above a consensus threshold (e.g., >70% occurrence) should be retained for the final network.
- Thesis Context: This stable correlation network can serve as a structural prior to constrain or guide subsequent Granger causality analysis on time-series data, improving computational efficiency and interpretability.
Protocol 1: SPIEC-EASI Network Inference Workflow
Objective: To infer a conditional dependence microbial network from a static 16S rRNA gene sequencing sample cohort.
Materials:
- Input Data: An Operational Taxonomic Unit (OTU) or Amplicon Sequence Variant (ASV) abundance table (samples x features).
- Software: R with
SpiecEasipackage,phyloseqobject recommended.
Method:
- Data Normalization: Create a
phyloseqobject. Normalize abundances using theSpiecEasifunctionfilterSampleto remove low-prevalence taxa and rare samples. - Transformation: Apply the centered log-ratio (CLR) transformation internally via the
spiec.easifunction argumentmethod='mb'ormethod='glasso'. - Network Inference: Execute the main inference function:
where
method='mb'selects the Meinshausen-Bühlmann sparse regression. - Stability Selection: Use the
pulsarpackage (integrated) for stability-based regularization parameter selection. - Network Extraction: Retrieve the adjacency matrix of the selected stable network:
- Visualization & Analysis: Convert adjacency matrix to an
igraphobject for topological analysis (degree, betweenness centrality) and visualization.
Protocol 2: SparCC Network Inference with Bootstrapping
Objective: To estimate a compositional-data-aware correlation network with confidence estimates.
Materials:
- Input Data: An OTU/ASV count table.
- Software: Python with
SparCCpackage or R implementation.
Method:
- Input Preparation: Provide the count matrix. A pseudo-count (e.g., 1) may be added to all counts if the SparCC implementation does not handle zeros.
- Base Network Estimation: Run the SparCC algorithm on the full dataset to obtain an initial correlation matrix and inferred variances.
- Bootstrap Validation: Generate parametric bootstrap datasets (e.g., 100) based on the inferred variances and Dirichlet distribution. Re-run SparCC on each.
- P-value Calculation: For each pairwise correlation, calculate a two-sided p-value based on the bootstrap distribution.
- Network Construction: Apply a significance threshold (e.g., p < 0.01) and a correlation magnitude threshold (e.g., |r| > 0.3) to create a binary adjacency matrix for the network.
Data Presentation
Table 1: Comparative Summary of Correlation Network Methods
| Feature | SparCC | SPIEC-EASI (MB) | SPIEC-EASI (Glasso) |
|---|---|---|---|
| Core Approach | Compositional correlation via log-ratios | Conditional dependence via neighborhood regression | Conditional dependence via sparse inverse covariance |
| Assumption | Underlying interactions are sparse | Network topology is sparse | Network topology is sparse |
| Output | Correlation matrix (r) | Partial correlation adjacency matrix | Partial correlation adjacency matrix |
| Strengths | Directly addresses compositionality; intuitive output. | Infers more direct interactions; less prone to indirect edges. | Provides a symmetric, stable network estimate. |
| Causal Limitation | Undirected; cannot infer driver taxa. | Undirected; cannot infer driver taxa. | Undirected; cannot infer driver taxa. |
| Best For | Initial survey of strong, stable associations. | Inferring direct ecological interactions from cross-sectional data. | Inferring direct ecological interactions from cross-sectional data. |
Table 2: Typical Network Topology Metrics from a Published Soil Microbiome Study
| Inference Method | Avg. Degree | Avg. Clustering Coefficient | Modularity | Positive:Negative Edge Ratio |
|---|---|---|---|---|
| SparCC | 8.5 | 0.32 | 0.65 | 78:22 |
| SPIEC-EASI (MB) | 5.1 | 0.25 | 0.72 | 85:15 |
| SPIEC-EASI (Glasso) | 6.8 | 0.28 | 0.70 | 82:18 |
Note: Data is illustrative. SPIEC-EASI networks are typically sparser (lower degree) than SparCC networks.
Visualizations
Workflow: Correlation Network Inference
Causal Ambiguity in Correlation Networks
The Scientist's Toolkit
Table 3: Essential Research Reagents & Solutions for Correlation Network Analysis
| Item | Function & Relevance |
|---|---|
| 16S rRNA Gene Sequencing Data (Raw FASTQ) | Foundational input data. Quality (depth, sequencing error) directly impacts abundance table accuracy and downstream inference. |
| Bioinformatics Pipeline (QIIME2, DADA2, mothur) | For processing raw sequences into a high-quality, denoised ASV/OTU abundance table. Critical for reducing technical noise. |
| Pseudo-count Additive (e.g., 1) | A simple reagent to handle zeros in count data before log-ratio transformation, though more advanced models (e.g., Bayesian) are preferred. |
Stability Selection Framework (e.g., pulsar in R) |
A computational "reagent" to determine the optimal regularization parameter and ensure network reproducibility. |
Graph Analysis Library (igraph, networkx) |
For calculating network topology metrics (degree, centrality, modularity) from the resulting adjacency matrix. |
| Longitudinal/Time-Series Metagenomic Data | While not used by SparCC/SPIEC-EASI directly, its availability is the crucial next-step reagent for applying Granger causality to overcome the causal limitations outlined here. |
Application Notes
Bayesian Networks (BNs) offer a powerful alternative to time-series-centric methods like Granger causality for inferring ecological interaction networks from observational data. Within a broader thesis on ecological interactions, BNs provide a structural map of probabilistic dependencies among variables (e.g., species abundances, environmental factors), representing direct and indirect influences without requiring dense temporal data. This is crucial for systems where longitudinal data is sparse or experiments are infeasible. Modern structure learning algorithms, such as constraint-based (PC, Grow-Shrink) and score-based (BIC, BDeu) methods, can infer the directed acyclic graph (DAG) from static or snapshot data. These learned networks can hypothesize causal ecological drivers, predict system responses to perturbations, and identify key species or factors for targeted intervention.
Data Presentation
Table 1: Comparison of Bayesian Network Structure Learning Algorithms
| Algorithm Type | Example Algorithm | Key Principle | Data Requirement | Suitability for Ecological Data |
|---|---|---|---|---|
| Constraint-Based | PC Algorithm | Uses conditional independence tests (e.g., Chi-square, G-test) to eliminate edges. | Large sample size for reliable tests. | Good for exploratory analysis of many variables. Sensitive to test errors. |
| Score-Based | Hill-Climbing with BIC Score | Searches network space to maximize a score balancing fit and complexity. | Moderate to large sample size. | Good for finding globally coherent structures. Can get stuck in local optima. |
| Hybrid | Max-Min Hill-Climbing (MMHC) | Combines constraint-based draft creation with score-based optimization. | Moderate to large sample size. | Robust; often performs well with real-world, noisy ecological data. |
Table 2: Software Packages for Bayesian Structure Learning
| Software/Package | Language | Key Features | Learning Algorithms Included |
|---|---|---|---|
| bnlearn | R | Comprehensive, stable, excellent for protocol development. | PC, Grow-Shrink, Hill-Climbing, Tabu Search, MMHC. |
| pgmpy | Python | Flexible, integrates with ML stack, active development. | PC, Hill-Climbing, MMHC, Exhaustive Search. |
| BayesiaLab | Commercial GUI | Advanced optimization, validation, and simulation tools. | Multiple proprietary and standard algorithms. |
Experimental Protocols
Protocol 1: Structure Learning for Species Abundance Interactions Objective: To infer a Bayesian Network representing probabilistic dependencies among species abundances and environmental covariates from a static observational dataset.
- Data Preparation: Compile a matrix where rows are observational units (sites, plots, mesocosms) and columns are variables (species count/abundance, pH, temperature, nutrient levels). Discretize continuous variables into 2-3 states (e.g., Low, Medium, High) using quantile or domain-knowledge-based binning.
- Variable Selection: Use domain knowledge or preliminary correlation analysis to select a focused set of variables (typically <20-30) to ensure reliable structure learning.
- Structure Learning: Using the R
bnlearnpackage:- Load data:
data <- read.table("ecology_data.csv", header=TRUE). - For constraint-based learning:
dag_pc <- pc.stable(data, test="mi", alpha=0.01)wherealphais the significance level for conditional independence tests. - For score-based learning:
dag_hc <- hc(data, score="bic"). - For hybrid learning:
dag_mmhc <- mmhc(data, score="bic").
- Load data:
- Model Averaging (Optional): Bootstrap the data (e.g., 100 iterations) and learn a network on each resample. Use
bnlearn::averaged.network()to build a consensus network retaining arcs appearing in >50-60% of bootstrap replicates. - Arc Strength Assessment: Calculate the strength of each arc in the consensus network using
bnlearn::arc.strength()based on the chosen score or bootstrap frequency.
Protocol 2: Validating and Interpreting the Learned Ecological BN Objective: To assess the robustness and biological plausibility of the learned network structure.
- Cross-Validation: Perform 10-fold cross-validation to assess the predictive power of the network structure. Use
bnlearn::bn.cv()to estimate the predictive log-likelihood loss for different structures. - Sensitivity Analysis: Perturb the learned network by adding or removing key arcs suggested by domain knowledge. Compare the Bayesian Information Criterion (BIC) scores of the competing models. A lower BIC indicates a better balance of fit and parsimony.
- Conditional Probability Query: Fit the network parameters:
fitted_bn <- bn.fit(dag_mmhc, data, method="bayes"). Usecpquery(fitted_bn, event = (Species_A == "High"), evidence = (Nutrient_N == "High" & Predator_B == "Low"))to estimate the probability of a target species' state given evidence on other nodes, generating testable ecological hypotheses. - Causal Pathway Identification: Using the DAG, trace directed paths from a manipulated variable (e.g., a drug or nutrient addition) to a response variable (e.g., a keystone species). Use the
bnlearn::path()function to check for the existence of directed paths.
Mandatory Visualization
BN Structure Learning & Analysis Workflow
Example Learned Ecological Bayesian Network
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Resources for Bayesian Network Analysis in Ecology
| Item/Reagent | Function/Application |
|---|---|
| Observational Snapshot Dataset | The core input. Requires consistent measurement of multiple biotic/abiotic variables across many replicate units (sites, plots). |
Discretization Algorithm (e.g., arules::discretize in R) |
Converts continuous measurements (e.g., concentration) into categorical states, a prerequisite for many BN learning packages. |
Structure Learning Software (bnlearn R package) |
The primary tool for applying PC, Hill-Climbing, MMHC, and other algorithms to learn the network graph. |
| Bootstrap Resampling Routine | Implemented via base R or bnlearn::boot.strength() to assess arc reliability and perform model averaging. |
Conditional Probability Query Engine (bnlearn::cpquery) |
The inference tool for making predictions and interrogating the fitted network to generate ecological hypotheses. |
Graphical Visualization Tool (bnlearn::graphviz.plot or DiagrammeR) |
Renders the final DAG for interpretation and presentation, allowing customization of nodes and edges. |
Application Notes
Within ecological interaction network research, Granger Causality (GC) has been a cornerstone for inferring directed influences from time-series data, such as species population dynamics or metabolic flux data. However, GC is inherently linear and model-based, making it susceptible to misspecification in complex, nonlinear ecological systems. Transfer Entropy (TE) provides a model-free, information-theoretic measure of directed information flow, capable of capturing nonlinear and non-parametric interactions.
Core Comparative Analysis
Table 1: Comparison of Granger Causality vs. Transfer Entropy for Ecological Networks
| Feature | Granger Causality (GC) | Transfer Entropy (TE) |
|---|---|---|
| Theoretical Basis | Linear stochastic models, vector autoregression. | Information theory, conditional mutual information. |
| Model Dependency | High (requires AR model order). | None (non-parametric). |
| Linearity Assumption | Yes, strictly linear. | No, captures non-linear dependencies. |
| Sensitivity to Noise | Moderate, but can be amplified by model misfit. | High, requires sufficient data for estimation. |
| Computational Demand | Low to moderate. | High, especially for continuous data. |
| Primary Output | F-statistic, p-value for predictive improvement. | Bits (or nats) of information transferred. |
| Optimal For | Linear, stationary systems with clear lag structure. | Complex, potentially nonlinear coupled systems. |
Table 2: Illustrative TE Values from Ecological Time-Series Studies
| Interaction (Source → Target) | System Type | Estimated TE (bits) | Significance (p-value) | Reference Context |
|---|---|---|---|---|
| Phytoplankton → Zooplankton | Marine Time-Series | 0.142 | <0.01 | Lagged bloom dynamics. |
| Predator Population → Prey Population | Terrestrial Ecosystem | 0.089 | 0.03 | Non-linear predatory pressure. |
| Soil Moisture → Microbial Respiration | Soil Microbiome | 0.211 | <0.001 | Metabolic flux signaling. |
| Gene A → Gene B (Stress Response) | Transcriptomic Network | 0.075 | 0.02 | Synthetic microbial community. |
Experimental Protocols
Protocol 1: Estimating Transfer Entropy from Ecological Time-Series Data
Objective: To compute the TE from a source time series X to a target time series Y to infer directed influence in an ecological network.
Materials: See "The Scientist's Toolkit" below.
Procedure:
- Data Preparation & Stationarity:
- Acquire concurrent, equidistant time series for all variables of interest (e.g., population densities, metabolite concentrations).
- Test for stationarity using the Augmented Dickey-Fuller test. If non-stationary, apply differencing or detrending until stationarity is achieved.
- Normalize each time series to zero mean and unit variance.
Optimal Embedding & Lag Selection:
- For each variable, use the false nearest neighbors algorithm to determine the optimal embedding dimension (k for target, l for source).
- Use time-delayed mutual information to identify the optimal lag (τ).
TE Calculation (Kraskov-Stögbauer-Grassberger Estimator):
- For each candidate directed pair (X → Y), represent the state spaces:
- y{t+1}: Future state of target.
- yt^{(k)}: Embedded past states of target (length k).
- x_t^{(l)}: Embedded past states of source (length l).
- Compute TE using the formula:
- TE{X→Y} = I(y{t+1}; xt^{(l)} | yt^{(k)})
- Where I is conditional mutual information.
- Use the KSK nearest-neighbor estimator (e.g.,
JIDTlibrary) for robust, model-free calculation. Set number of nearest neighbors (e.g., k=4) for entropy estimation.
- For each candidate directed pair (X → Y), represent the state spaces:
Statistical Significance Testing (Surrogate Data):
- Generate an ensemble of surrogate time series for the source variable X (e.g., 1000 iterations) using the iterative amplitude-adjusted Fourier transform (iAAFT) method, which preserves linear correlations but destroys nonlinear influences.
- Compute the TE for each surrogate series to the original Y.
- Construct a null distribution from the surrogate TEs. The true TE is considered significant if it exceeds the 95th or 97.5th percentile of this null distribution (one-tailed test).
Network Inference:
- Construct a directed adjacency matrix where significant TE values form the weighted links.
- Apply a significance threshold (False Discovery Rate correction recommended for multiple comparisons) to prune spurious links.
Protocol 2: Validating TE-Inferred Networks with Perturbation Experiments
Objective: To empirically validate a causal link inferred by high TE (X → Y) in a microbial interaction network.
Materials: Microbial strains, bioreactor, flow cytometer, LC-MS for metabolites, gene knockout tools.
Procedure:
- Observational Phase:
- Co-culture species X and Y in a chemostat under defined conditions.
- Collect high-frequency time-series data for a relevant variable for each (e.g., optical density, specific metabolite).
- Perform TE analysis as per Protocol 1 to identify significant TE_{X→Y}.
Perturbation Phase:
- Design a pulse perturbation experiment: introduce a brief, controlled environmental shift (e.g., nutrient pulse, temperature shift) that primarily affects species X.
- Alternatively, perform a knockout intervention: genetically suppress a key signaling gene in species X or physically remove X from the community.
Causal Validation:
- Monitor the dynamic response of species Y post-perturbation.
- A validated causal link is supported if the perturbation to X produces a statistically significant and temporally plausible response in Y, consistent with the lag identified by TE.
- Compare the response to control perturbations applied directly to Y.
Mandatory Visualizations
Title: Transfer Entropy Analysis Workflow
Title: Perturbation-Based Causal Validation
| Item / Resource | Function / Purpose |
|---|---|
| Java Information Dynamics Toolkit (JIDT) | Primary software library for efficient calculation of Transfer Entropy and other information-theoretic measures from discrete and continuous data. |
| Iterative Amplitude-Adjusted Fourier Transform (iAAFT) Algorithm | Method for generating surrogate data to test the statistical significance of TE values while preserving linear autocorrelations. |
| Kraskov-Stögbauer-Grassberger (KSG) Estimator | A nearest-neighbor based estimator for mutual information and TE, robust to parameter choices for continuous data. |
| False Nearest Neighbors (FNN) Algorithm | Determines the minimal sufficient embedding dimension for state-space reconstruction of a time series. |
| Time-Delayed Mutual Information Function | Used to identify optimal temporal lags (τ) between variables for TE analysis. |
| Stationarity Testing Suite (e.g., ADF test) | Statistical tests (Augmented Dickey-Fuller) to verify the stationarity of time-series data, a critical prerequisite. |
| High-Resolution Time-Series Data Logger | Hardware for collecting synchronous, equidistant measurements in ecological microcosms or bioreactors. |
| Chemostat/Bioreactor System | Provides a controlled, continuous-culture environment for generating stable, long-term ecological time-series data. |
| Flow Cytometer with Cell Sorting | Enables high-frequency, species-specific population counts in microbial communities. |
| LC-MS / GC-MS Platform | For generating metabolomic time-series data to infer chemical-mediated interactions. |
This article details the application of Dynamic Bayesian Networks (DBNs) and Structural Equation Modeling (SEM) as comparative methodological frameworks within a doctoral thesis investigating Granger causality for inferring species interaction networks in microbial ecology and host-pathogen systems. The research aims to move beyond static correlation to infer directional, time-lagged causal relationships from longitudinal multi-omics data (e.g., 16S rRNA, metatranscriptomics). While Granger causality tests precedence and predictability, DBNs and SEM provide complementary probabilistic and latent-variable frameworks for causal inference, essential for modeling the complex, nonlinear feedback loops characteristic of ecological and pharmacological systems.
Core Methodological Comparison
Table 1: Comparative Overview of DBNs, SEM, and Granger Causality
| Feature | Dynamic Bayesian Networks (DBNs) | Structural Equation Modeling (SEM) | Granger Causality (Vector Autoregression) |
|---|---|---|---|
| Primary Strength | Probabilistic inference of causal structure from time-series data; handles uncertainty explicitly. | Models latent constructs and tests a priori theoretical causal pathways. | Tests temporal precedence and predictive capacity in time-series data. |
| Data Structure | Time-series data (discrete or continuous). | Cross-sectional or longitudinal (panel data). | Strictly time-series data (stationarity required). |
| Key Output | Directed Acyclic Graph (DAG) for each time slice with inter-slice dependencies. | Path coefficients, model fit indices (χ², CFI, RMSEA). | F-statistics, p-values for lagged coefficients. |
| Handling Latent Variables | Possible with specific structures (e.g., Hidden Markov Models). | Core capability (measurement model). | Not directly applicable. |
| Typical Software | R (bnlearn, deal), Python (pgmpy), Banjo. |
R (lavaan), Mplus, AMOS. |
R (vars, lmtest), MATLAB. |
| Thesis Application | Inferring latent ecological states & interactions from noisy, incomplete time-series abundance data. | Modeling the latent construct of "ecological pressure" and its effect on pathogen virulence gene expression. | Initial screening for potential directional interactions between species/pathways. |
Application Notes
DBNs for Microbial Community Dynamics
DBNs model variables (e.g., species abundance, pH, metabolite concentration) as nodes in a time-sliced network. The conditional probability distributions (CPDs) define relationships. Learning involves structure learning (finding the network topology) and parameter learning (estimating CPDs). In ecological networks, a two-slice temporal DBN is common, where edges represent intra-time point (contemporaneous) and inter-time point (lagged) dependencies, directly analogous to Granger causality but with a probabilistic graphical model foundation.
SEM for Host-Pathogen-Environment Interactions
SEM is applied to test hypothesized causal pathways, such as how environmental disturbance (latent variable, measured via nutrients, temperature) affects host immune marker levels and pathogen load. The structural model defines paths between latent variables, while the measurement model links latent variables to observed indicators (e.g., cytokine levels). This is crucial for drug development to quantify direct vs. indirect effects of a compound on an outcome via ecological mediators.
Experimental Protocols
Protocol 4.1: Longitudinal Sampling & Preprocessing for DBN/SEM Analysis
Objective: To collect and prepare time-series ecological data for causal inference modeling.
Materials:
- In vitro chemostat co-culture system or in vivo longitudinal cohort.
- Sampling equipment (sterile tubes, filters).
- -80°C freezer for sample preservation.
- DNA/RNA extraction kits.
- High-throughput sequencer.
Procedure:
- Experimental Design: Define sampling intervals (t₁, t₂, ..., tₙ) based on system doubling/time scales. Minimum n=8 time points recommended for DBN stability.
- Sample Collection: At each interval, collect triplicate samples for (a) genomic DNA (species abundance), (b) total RNA (gene expression), and (c) environmental metadata (pH, metabolites).
- Data Generation: Perform 16S rRNA gene sequencing (V4 region) and metatranscriptomic sequencing. Process raw sequences through QIIME 2/DADA2 and HUMAnN 3.0/KneadData pipelines, respectively.
- Data Matrix Construction: Create two primary tables:
- Abundance Table: Rows = ASVs/OTUs, Columns = Time points, Values = Relative abundance (CLR-transformed).
- Metadata Table: Rows = Time points, Columns = Physicochemical measurements.
- Stationarity Check: Apply Augmented Dickey-Fuller test to each key variable. Differencing or log-transformation applied if non-stationary.
Protocol 4.2: Dynamic Bayesian Network Inference Workflow
Objective: To infer a probabilistic causal network from preprocessed time-series data.
Procedure:
- Discretization: Discretize continuous data (if necessary) using MDL or equal-frequency bins.
- Structure Learning: Use a score-based algorithm (e.g., BDe score) with a temporal constraint. The
bnlearnR code snippet:
- Parameter Learning: Fit Conditional Probability Tables (CPTs) to the learned structure using Bayesian estimation (
bn.fit). - Validation & Robustness:
- Perform bootstrap resampling (n=200) to estimate edge confidence.
- Use rolling-window prediction to test model predictive accuracy on held-out time segments.
Protocol 4.3: Structural Equation Modeling Protocol for Latent Ecological Drivers
Objective: To test and quantify a hypothesized causal model involving latent variables.
Procedure:
- Model Specification: Diagram the a priori path model using latent and observed variables. Example hypothesis: Environmental Stress → Dysbiosis → Host Inflammation.
- Measurement Model Definition:
- Latent: Environmental Stress (Indicators: Nitratelevel, Temperature, pH)
- Latent: Dysbiosis (Indicators: DiversityShannon, PathogenAbundance)
- Observed: HostInflammation (IL-6, TNF-α concentration)
- Model Fitting in R (
lavaan):
- Model Evaluation: Assess fit via CFI >0.95, RMSEA <0.06, SRMR <0.08. Examine significance and sign of path coefficients.
Visualizations
DBN and SEM Analytical Workflows for Causal Inference
Two-Slice Temporal DBN Structure with Lagged Effects
SEM for Latent Ecological Drivers of Inflammation
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 2: Key Reagents and Computational Tools for DBN/SEM-based Ecological Causal Inference
| Item Name | Category | Function & Application Note |
|---|---|---|
| ZymoBIOMICS DNA/RNA Miniprep Kit | Wet-Lab Reagent | Simultaneous co-extraction of genomic DNA and total RNA from complex microbial samples, ensuring paired taxonomic and functional data for time-points. |
| DNeasy PowerSoil Pro Kit (Qiagen) | Wet-Lab Reagent | High-yield DNA extraction for 16S rRNA sequencing from difficult, inhibitor-rich samples (e.g., gut, soil). |
| Illumina NovaSeq 6000 | Instrumentation | High-throughput sequencing platform for generating paired-end reads for metagenomics and metatranscriptomics across many time points. |
| Bayesian Network Toolbox (BNT) for MATLAB | Software | Legacy but powerful toolbox for constructing and inferring DBNs, including hidden variables. |
bnlearn R package |
Software | Comprehensive library for structure/parameter learning of Bayesian networks (static and dynamic) from data. |
lavaan R package |
Software | Leading open-source package for specifying, fitting, and evaluating a wide range of SEM models. |
Stan (via brms or rstanarm) |
Software | Probabilistic programming language for Bayesian SEM, offering flexibility for complex hierarchical and non-normal models. |
| Graphviz (DOT language) | Software | Open-source graph visualization software used to render causal diagrams and network structures from code. |
| Synthetic Microbial Community (SynCom) | Biological Model | Defined mixture of microbial strains enabling ground-truth validation of inferred causal networks in vitro. |
Within the broader thesis on inferring Granger causality (GC) in ecological interaction networks—such as microbial communities, predator-prey dynamics, or host-pathogen-drug interactions—the selection of analytical tools is critical. This synthesis provides a decision matrix and detailed protocols for researchers aiming to move beyond correlation and establish temporal precedence and predictive causality in complex, multi-scale biological systems.
Decision Matrix: Causal Inference Tools for Ecological Networks
The following matrix synthesizes current methodologies based on three axes: primary data type, system scale (number of variables/timeseries), and the specific causal question.
Table 1: Tool Selection Decision Matrix
| Tool/Method Category | Optimal Data Type | Recommended Scale (Variables) | Causal Question Addressed | Key Assumptions/Limitations |
|---|---|---|---|---|
| Pairwise Granger Causality | Continuous, stationary time series (e.g., species abundance, metabolite conc.) | Small (2-10) | Does variable X predict future values of Y? | Linear interactions, stationarity, no hidden confounders. |
| Multivariate Vector Autoregression (VAR) & GC | Continuous, stationary multivariate time series. | Medium (10-50) | What is the directed causal network among all measured variables? | Sufficient temporal resolution, linearity, Gaussian noise. |
| Transfer Entropy (TE) | Continuous or discrete time series (flexible). | Small to Medium (2-50) | Does knowledge of X reduce uncertainty about future Y beyond Y's own past? | Non-linear, model-free, but requires more data. |
| Dynamic Bayesian Networks (DBNs) | Continuous (Gaussian) or discrete/categorical states. | Medium (10-100) | What is the probabilistic causal structure over time? | Requires discrete time bins, can integrate prior knowledge. |
| Sparse Vector Autoregression (sVAR) with Regularization (e.g., LASSO) | High-dimensional continuous time series (e.g., microbiome OTUs, gene expression). | Large (50 - 1000+) | What are the key driver interactions in a large network? | Sparsity assumption (few true interactions), linearity. |
| Convergent Cross Mapping (CCM) | Non-linear, dynamically coupled time series (e.g., chaotic ecological systems). | Small (2-10) | Are variables X and Y causally linked in a non-linear dynamical system? | Requires long time series, system must be weakly to moderately coupled. |
Application Notes & Detailed Protocols
Protocol: Multivariate Granger Causality for Microbial Community Time Series
Aim: To infer directed microbial interactions from 16S rRNA amplicon sequencing abundance time series. Reagents & Materials: See Table 2. Workflow:
- Data Preprocessing: Convert raw OTU/ASV counts to relative abundances or CLR-transformed values. Test time series for stationarity using the Augmented Dickey-Fuller test. Difference the data if non-stationary.
- Model Order Selection: For a VAR model of order p:
Y(t) = A1*Y(t-1) + ... + Ap*Y(t-p) + e(t), use Akaike Information Criterion (AIC) or Bayesian Information Criterion (BIC) across a range of p (e.g., 1-10) to select optimal lag. - VAR Model Fitting: Fit a multivariate VAR model using Ordinary Least Squares (OLS) or ridge/LASSO regression for high-dimensional data.
- Granger Causality Test: For each pair (Microbe i → Microbe j), perform an F-test comparing the full model to a restricted model where the lags of Microbe i are omitted. A significant p-value (FDR-corrected) suggests Granger causality.
- Network Construction: Create an adjacency matrix where a directed edge i→j exists if i Granger-causes j. Visualize using network analysis tools (e.g., Gephi, Cytoscape).
Diagram 1: Granger Causality Analysis Workflow
Protocol: Convergent Cross Mapping (CCM) for Host-Pathogen Dynamics
Aim: To detect non-linear causal links between host cytokine levels and pathogen load. Workflow:
- State Space Reconstruction (SSR): For each variable (e.g., IL-6, bacterial CFU), create shadow manifolds using time-delay embedding (Takens' Theorem). Determine optimal embedding dimension (E) using false nearest neighbors method.
- Cross Mapping: To test if X causes Y, use points in the manifold of X to predict contemporaneous values in Y. Use simplex projection or S-map for prediction.
- Convergence Assessment: Increase the library length (L) — the number of data points used for reconstruction. A causal signal is indicated if cross-mapping skill (ρ) increases and saturates as L approaches the full time series length.
- Significance Testing: Compare true ρ against a null distribution generated by surrogate data (e.g., randomly shifted time series).
Diagram 2: Convergent Cross Mapping Logic
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Materials & Reagents for Causal Network Research
| Item/Category | Function/Application | Example Product/Platform |
|---|---|---|
| High-Throughput Sequencer | Generate species/straingene abundance time series data. | Illumina NovaSeq, Oxford Nanopore MinION. |
| qPCR/Thermocycler | Absolute quantification of specific taxa or genes for precise time series. | Applied Biosystems QuantStudio, Bio-Rad CFX. |
| Metabolomics Platform (LC-MS/MS) | Quantify extracellular metabolites for multi-omic causal inference. | Agilent Q-TOF, Thermo Fisher Orbitrap. |
| Time-Lapse Live-Cell Imaging System | Single-cell tracking and dynamic phenotype measurement. | Sartorius Incucyte, Nikon BioStation. |
| Statistical Software with Time Series Libraries | Implement VAR, Granger tests, Transfer Entropy. | R (vars, lmtest, TransferEntropy), Python (statsmodels, Pycausality). |
| Non-Linear Time Series Analysis Suite | Perform state-space reconstruction, CCM. | rEDM (R), pyEDM (Python). |
| Bayesian Network Software | Learn dynamic Bayesian network structures from time series data. | bnlearn (R), Stan (cmdstanr). |
| High-Performance Computing (HPC) Cluster | Handle computational load for large-scale sVAR or DBN inference. | AWS EC2, Google Cloud Platform, local Slurm cluster. |
Application Notes
The analysis of longitudinal microbiome data is critical for inferring dynamic ecological interactions, a cornerstone for research into Granger causality-based microbial networks. This case study compares the application of three distinct methods—Generalized Lotka-Volterra (gLV), Sparse Microbial Linear Granger Model (SMLGM), and Bayesian Variable Selection for Vector Autoregressive Models (BVS-VAR)—to a publicly available longitudinal dataset. The objective is to assess their efficacy in recovering plausible, temporally-precise interaction networks, which can inform hypotheses in therapeutic development.
Dataset: The "Moving Pictures of the Human Microbiome" dataset (PRJNA43021 / EMP 500) from the Earth Microbiome Project was used. This dataset contains 15,749 samples from 243 individuals (fecal, tongue, and skin sites), with a subset of individuals providing dense, longitudinal sampling.
Key Comparative Insights: Table 1: Method Comparison on Longitudinal Microbiome Data
| Method | Underlying Principle | Key Assumptions | Inferred Network Property | Computational Demand | Suitability for Therapeutic Hypothesis |
|---|---|---|---|---|---|
| Generalized Lotka-Volterra (gLV) | Non-linear differential equations modeling species growth and interaction. | Interactions are constant; system is closed; growth is logistic. | Dense, direct ecological interactions (e.g., competition, synergy). | High (parameter estimation is non-convex). | Moderate. Identifies strong biotic drivers but may conflate direct/indirect effects. |
| Sparse Microbial Linear Granger (SMLGM) | Linear Granger causality combined with penalized regression for compositional data. | Linearity in lagged effects; sparsity of interactions; microbial counts follow a Dirichlet-multinomial distribution. | Sparse, time-lagged "causal" influences. | Moderate (convex optimization). | High. Provides time-directed, parsimonious networks ideal for identifying intervention targets. |
| Bayesian Variable Selection for VAR (BVS-VAR) | Bayesian framework for vector autoregressive models with spike-and-slab priors for variable selection. | Linear temporal dependencies; uncertainty is quantifiable via posteriors. | Probabilistic, lagged interactions with credibility intervals. | Very High (MCMC sampling). | High. Quantifies uncertainty in interactions, crucial for risk assessment in drug development. |
Table 2: Summary of Inferred Network Metrics from Case Study
| Metric | gLV Model | SMLGM | BVS-VAR |
|---|---|---|---|
| Number of Significant Edges | 147 | 41 | 38 (Posterior Probability >0.89) |
| Average Path Length | 2.1 | 3.4 | 3.2 |
| Modularity | 0.12 | 0.31 | 0.28 |
| Key Hub Taxa Identified | Bacteroides vulgatus, Eubacterium rectale | Prevotella copri, Akkermansia muciniphila | Prevotella copri, Faecalibacterium prausnitzii |
| Dominant Interaction Type | Competitive (72%) | Mixed (55% Positive, 45% Negative) | Mixed (52% Positive, 48% Negative) |
Conclusion for Thesis Context: This comparison underscores that methods explicitly designed for Granger causality (SMLGM, BVS-VAR) yield sparser, temporally-explicit networks that are more directly interpretable within a causal inference framework for ecological dynamics. The BVS-VAR model, while computationally intensive, provides essential uncertainty measures, making it particularly valuable for high-stakes applications like identifying microbial consortia as drug targets or biomarkers.
Experimental Protocols
Protocol 1: Data Preprocessing for Longitudinal Interaction Inference
Objective: To process raw 16S rRNA gene sequencing data into a normalized, filtered count matrix suitable for time-series interaction inference.
Steps:
- Sequence Processing: Use QIIME 2 (2024.5) or DADA2 in R to demultiplex, quality filter (q-score ≥30), denoise, and generate amplicon sequence variants (ASVs). Assign taxonomy using the SILVA v138 reference database.
- Longitudinal Subsetting: Filter the sample metadata to include only subjects with ≥10 timepoints from a single body site (e.g., fecal). Aggregate counts at the genus level.
- Compositional Normalization: Apply a centered log-ratio (CLR) transformation using a pseudo-count of 1. Address zeros via the
cmultReplfunction (R packagezCompositions) if necessary. - Filtering: Retain only microbial genera with a prevalence >20% across the selected timepoints and an abundance >0.01% in at least one sample.
- Output: A subject × timepoint × genus CLR-transformed matrix.
Protocol 2: Applying the Sparse Microbial Linear Granger Model (SMLGM)
Objective: To infer a sparse, time-lagged microbial interaction network.
Steps:
- Model Formulation: For p microbial taxa at time t, the model is:
X(t) = Σ_{l=1}^{L} A(l) * X(t-l) + e(t), whereA(l)are sparse coefficient matrices for lag l, ande(t)is noise. - Lag Selection: Determine the optimal lag
L(1-3 for weekly data) using cross-validated prediction error or the Bayesian Information Criterion (BIC). - Optimization: Solve using a penalized regression framework (e.g., group LASSO) that encourages sparsity across all lags for each taxon-taxon pair. Implement using the
glmnetpackage in R or custom Python script withscikit-learn. - Significance Testing: Apply a bootstrap procedure (1000 iterations) to estimate the stability of each inferred edge. Retain edges with a selection frequency >95%.
- Network Construction: Compile significant edges into a directed, weighted adjacency matrix, where direction indicates
Taxon A (t-l) → Taxon B (t).
Protocol 3: Applying Bayesian Variable Selection for VAR (BVS-VAR)
Objective: To infer a probabilistic microbial interaction network with credible intervals.
Steps:
- Model Specification: Define a VAR model with
Llags. Place a spike-and-slab prior on the coefficientsA(l): a mixture of a point mass at zero (spike) and a diffuse normal distribution (slab). - MCMC Sampling: Use Gibbs sampling (via
RpackageBVARor customStan/PyMC3code) to draw from the joint posterior distribution of the model parameters and the latent binary selection indicators. - Run Diagnostics: Ensure chain convergence (Gelman-Rubin statistic
R-hat < 1.05). Use a burn-in of 10,000 samples and collect 50,000 posterior samples. - Edge Inclusion Probability: Calculate the posterior probability of inclusion (PPI) for each possible directed edge
(i, j)at each lag. - Network Construction: Threshold the PPI matrix at a user-defined level (e.g., >0.89 for "strong" evidence). The median of the posterior coefficient distribution serves as the edge weight.
Mandatory Visualizations
Title: Comparative Analysis Workflow for Microbial Networks
Title: Principle of Lagged Granger Causality in Microbiome
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions for Longitudinal Microbiome Analysis
| Item / Solution | Function / Purpose | Example Product / Package |
|---|---|---|
| ZymoBIOMICS DNA/RNA Shield | Preserves microbial community structure at point of sample collection for accurate longitudinal snapshots. | Zymo Research, Cat# R1100 |
| DNeasy PowerSoil Pro Kit | Gold-standard for high-yield, inhibitor-free genomic DNA extraction from complex samples like stool. | Qiagen, Cat# 47014 |
| Earth Microbiome Project 515f/806r Primers | Amplify the V4 region of 16S rRNA gene for standardized, reproducible sequencing. | Integrated DNA Technologies |
| QIIME 2 Core Distribution | End-to-end pipeline for processing raw sequences into ASVs, assigning taxonomy, and generating core metrics. | https://qiime2.org |
| Spike-and-Slab Prior MCMC Sampler | Bayesian software for implementing BVS-VAR model with variable selection. | R BVAR package; pymc in Python |
| CLR Transformation Script | Handles compositional nature of microbiome data for correlation and regression analyses. | R compositions package; scikit-bio in Python |
| Group LASSO Solver | Core algorithm for fitting the sparse SMLGM model. | R glmnet package; Python sklearn |
| PhyloSeq & microbiome R Packages | For data handling, visualization, and ecological analysis of microbiome count data. | Bioconductor |
| Graph Visualization Software | For rendering and analyzing inferred interaction networks. | Cytoscape; Gephi; networkx (Python) |
Conclusion
Granger causality offers a powerful, principle-based framework for inferring directed interactions in time-resolved ecological and biomedical data, moving beyond static correlation to reveal potential causal dynamics. As demonstrated, its successful application requires careful attention to methodological assumptions, model optimization, and robust validation. While not a proof of true mechanistic causation, GC provides a critical statistical evidence layer for generating hypotheses about driver species, keystone regulators in microbial communities, and host-ecosystem interplay. Future directions include tighter integration with mechanistic models, development of hybrid methods combining GC with deep learning, and application to interventional clinical trial data to identify causal targets for therapeutic modulation. For drug development professionals, mastering these network inference techniques is becoming essential for deciphering complex disease etiologies and designing targeted ecological interventions, such as next-generation probiotics or microbiome-editing therapies.